1 引言(Introduction)
在石油天然气开采、炼制、化工、冶金等行业生产过程中均会产生大量的含硫废水, 含硫废水中的硫化物具有毒性大、腐蚀性强等特点, 会严重破坏金属设备管线,给城市、工业污水系统及油气田开发带来巨大的经济损失.含硫废水还会溢出对神经系统具有强烈毒害作用且具有刺激性气味的H2S气体, 既危害人体健康, 又对环境造成极大污染.因此, 研究如何高效简单地治理此类废水成为人们关注的焦点.
目前, 国内外常采用混凝沉淀法、汽提法、氧化法等处理含硫废水, 而氧化法因快速直接且易于操作故在含硫废水处理方面倍受青睐.常用的氧化法通常是将硫化物直接氧化为高价态硫氧酸根, 但硫氧酸根易被硫酸盐还原菌再次还原成硫化物造成二次污染,若将硫化物直接转化成单质硫回收, 既能解决环境污染问题, 又能实现废水的资源化利用.目前, 能将废水中硫化物转化成单质硫的方法多为生物法和电化学法,而关于以单质硫为目标产物的化学氧化法的报道极少.由于生物法多适用于低浓度含硫废水, 且由微生物体内排出的微小硫颗粒在水中呈胶体状态, 难以自然沉淀;电化学法生成的单质硫会在阳极表面沉积, 需要定期将单质硫从电极上除去, 以免影响电池的正常工作(Paritam et al., 2010);化学氧化法能处理高浓度的含硫废水且生成的单质硫通过过滤即可获得.因此, 本研究选用过氧化氢为氧化剂, 通过控制反应体系氧化还原电位(ORP), 以实现将含硫废水中的硫化物定向氧化为单质硫的目标;同时, 研究氧化过程中单质硫的晶体结构和微观形貌并探讨单质硫颗粒增大的主要原因, 以期为实现硫化物定向氧化和单质硫资源化回收利用奠定理论基础.
2 材料与方法(Materials and methods) 2.1 实验材料
实验采用Na2S・9H2O配成质量浓度为2000 mg・L-1的模拟含硫废水, 实验中所用30%过氧化氢、聚丙烯酰胺、丙三醇、氢氧化钠、硫代硫酸钠、碘、盐酸等试剂均为分析纯.
2.2 实验装置与仪器
实验采用自制的氧化脱硫装置(图 1), 主要包括反应器和尾气吸收装置两个部分, 整个反应在密闭条件下进行, 并通过尾气吸收装置吸收反应过程中产生的H2S气体.实验所用仪器包括雷磁PHS-3E型pH计、雷磁213-10型铂电极、78-1型磁力加热搅拌器、SHB-Ⅲ型循环水式多用真空泵.
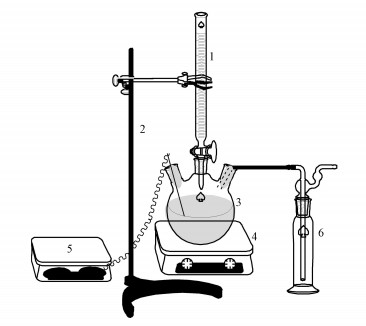
图 1 氧化脱硫装置(1.酸式滴定管2.铁架台3.三口圆底烧瓶4.磁力加热搅拌器5.pH计/铂电极6.尾气吸收瓶)
2.3 实验方法
取200 mL含硫废水于250 mL三口烧瓶中, 加入过氧化氢9 mL・L-1, 通过酸式滴定管加酸调节废水初始pH为6, 在50 r・min-1的转速下搅拌反应10 min, 并在反应过程中通过控制加酸量控制反应体系ORP, 反应结束后用0.45μm微孔滤膜过滤, 分别对滤液中可溶性产物及固相产物进行测定.在不同反应时间下对液相中单质硫进行扫描电镜分析和粒径测试.
2.4 分析方法
反应后滤液中SO42-采用重量法(GB11899―1989)测定, S2O32-采用滴定法测定, 体系ORP采用雷磁铂电极测定, 固相产物晶体结构采用X Pert PRO MPD型X射线衍射仪分析, 微观形貌采用JSM-7500F型扫描电镜分析, 颗粒粒径采用PALS/90PLUS型Zeta电位及粒度分析仪测量.氧化产物收率计算公式如下:
(1)
式中, Cx、C0分别代表氧化产物中硫元素的含量和原水中硫化物浓度(mg・L-1).
3 结果与讨论(Results and discussion) 3.1 受控氧化过程对单质硫收率的影响
由于硫元素具有多种价态, 因此, 在氧化还原反应中会产生多种氧化产物, 形成多个氧化还原电对, 通过控制反应体系氧化还原电位(ORP)可以控制氧化反应的进程(杨钙仁等, 2009).在过氧化氢投加量为9 mL・L-1、初始pH为6、反应时间为10 min的条件下, 不同ORP值对各氧化产物收率的影响如图 2所示.

图 2 体系ORP对氧化产物收率的影响
由图 2可知, 含硫废水受控氧化的产物包括单质硫(S0)、S2O32-和SO42-, 其中, 单质硫为主要氧化产物且收率受ORP影响较大, 随反应体系ORP值的升高先增大后减小, 在ORP为(30±5)mV时达到最大值76.35%, 而S2O32-的收率则随ORP值的升高而逐渐减小, 当OPR由(-50±5)mV升高至(50±5)mV时, S2O32-收率由26.54%减小至5.32%.受控氧化过程主要包括以下几个反应:
(2)
(3)
(4)
由以上反应式可知, 要对含硫废水进行受控氧化使单质硫成为主要产物, 应将反应(2)作为目标反应, 反应(3)、(4)则为副反应.根据标准电极电势列表及相关计算(宋天佑等, 2010)可知, 酸性条件下各电对的标准电极电势分别为:EAθ(H2O2/H2O)=1.776 V、EAθ(S/HS-)=-0.065 V、EAθ(SO42-/HS-)=0.251 V、EAθ(S2O32-/HS-)=0.20 V, 只有当氧化剂电对的电势大于还原剂电对的电势时才能发生氧化还原反应, 且两者的差值越大, 反应进行得越完全, 因此, 相对副反应(3)、(4), 目标反应(2)发生的趋势更大, 适当减小体系的ORP值使其接近副反应的电极电势有利于抑制副反应的发生, 从而更有利于目标反应的进行.
另一方面, 体系的pH值与ORP值的变化呈反向关系,其变化曲线如图 3所示.由图 3可知, 当体系ORP值为(30±5)mV时, 体系pH值维持在6.25~6.45之间;当体系ORP值由30 mV减小至-40 mV时, pH值由6.35增大至8.00致使体系呈碱性, 而碱性条件下过氧化氢的标准电极电势仅为0.867 V(宋天佑等, 2010), 同时还会发生分解, 使有效氧化组分减少, 从而不利于目标反应的进行.此外, 体系ORP值大于60 mV后会导致体系处于过酸的环境(pH<6), 此时HS-会进一步质子化生成H2S, 而H2S转化为单质硫的标准电极电势为0.142 V(宋天佑等, 2010), 比HS-转化为单质硫的标准电极电势大, 因此, 更不利于单质硫的形成.

图 3 体系pH值与ORP的关系
S2O32-收率随ORP值的升高而逐渐减小的主要原因在于体系ORP值越高, pH值越低, 当体系处于酸性条件时S2O32-会发生自氧化反应, 具体如式(5)所示.可知, S2O32-的自氧化反应会导致自身产量的降低, 同时使单质硫产量增加, 这也是单质硫收率先随ORP值的升高而逐渐增大的另一个原因.综上所述, 反应过程中将体系ORP控制在(30±5)mV时, 体系pH值维持在6.25~6.45之间, 恰好处于弱酸性条件, 既能保证过氧化氢的氧化能力又能使硫化物主要以HS-形式存在, 此时能较好实现含硫废水的受控氧化, 同时副产物产量得以有效控制, 单质硫收率达76.35%.
(5)
3.2 单质硫的晶体结构
将氧化反应所得固相产物在室温下风干并研磨后进行XRD分析, 所得的X射线衍射图谱与单质硫标准图谱的对比如图 4所示.由图 4可知, 过氧化氢氧化含硫废水过程中产生的固相产物的X射线衍射图谱与单质硫标准图谱基本一致, 说明体系中产生的固相产物是单质硫.图中强度较大的几个衍射峰分别在2θ为23.082°、25.879°、26.749°、27.769°和28.680°处出现, 与标准图谱的位置一致, 分别对应S的(222)、(026)、(311)、(040)和(313)晶面族.这说明过氧化氢氧化脱硫过程中产生的单质硫成分较纯, 杂质少.分子Sn可以由硫原子以多种不同的方式组合而成(如S2、S4、S6和S8等), 其主要存在形式为环状分子或链状分子(陈锋, 2013).在硫的环状分子中S8最稳定, S8由于在晶格中排列方式的不同又可形成斜方硫(α-硫)和单斜硫(β-硫), 其中, 只有斜方硫才能在室温下稳定存在.因此, 过氧化氢氧化处理含硫废水过程中产生的单质硫主要为S8环状分子、正交晶系斜方硫(郑伟, 2006).
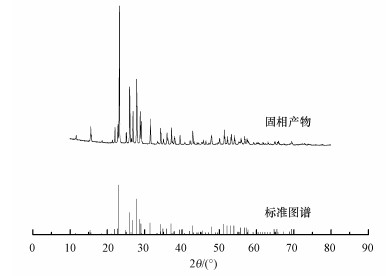
图 4 固相产物的X射线衍射图
3.3 单质硫的微观形貌
在不同反应时间(5 s、3 min、10 min)下, 对液相中单质硫进行扫描电镜分析, 结果如图 5所示.由图 5可知, 当反应时间为5 s时, 体系中的单质硫在放大倍数为1000倍下观察呈絮状(图 5a);当反应时间为3 min时, 体系中的单质硫呈薄层状, 且有向内部包裹的趋势(图 5b);当反应时间为10 min时, 体系中单质硫呈大小不一, 形状为不规则的颗粒状(图 5c).将不同时间下的单质硫放大到20000倍观察, 反应时间为5 s时, 体系中的单质硫是由无数个粒径极小的颗粒聚在一起形成的层状物, 其中较为规则的颗粒粒径约为237.5 nm(图 5d); 而随着时间的延长, 层状物逐渐向内包裹, 到反应时间为10 min时, 层状的单质硫变为粒径较大的不规则的颗粒硫, 其中相对规则的颗粒粒径达到了5.563μm(图 5f).结果表明, 在过氧化氢氧化含硫废水的最初阶段, 单质硫颗粒属于纳米颗粒, 反应一段时间后, 纳米级颗粒团聚在一起, 形成了微米级颗粒.其主要原因是纳米硫颗粒最初由S8环状分子结晶而成, 而形成的纳米硫颗粒由于粒度小, 比表面积大, 表面能大, 处于能量不稳定状态, 根据能量最低原理, 在没有其他附加条件的情况下, 纳米粒子会自发地团聚(冯拉俊等, 2004).另外, 在搅拌条件下, 体系中具有一定的离心力, 也会使体系中的纳米硫颗粒相互碰撞而发生聚沉.

图 5 液相中单质硫扫描电镜照片(a、b、c分别表示反应5 s、3 min、10 min, ×1000; d、e、f分别表示反应5 s、3 min、10 min, ×20000)
3.4 单质硫的生长团聚
由于单质硫颗粒大小直接影响单质硫在反应体系中的稳定性及沉降可分离性能, 其粒度越大越有利于单质硫的分离回收, 因此, 考察反应过程中单质硫颗粒粒度变化规律能为单质硫的有效分离和资源化回收利用提供理论支持.在不同反应时间(5 s、30 s、1 min、2 min、3 min)下, 液相中单质硫颗粒粒度变化规律如图 6所示.
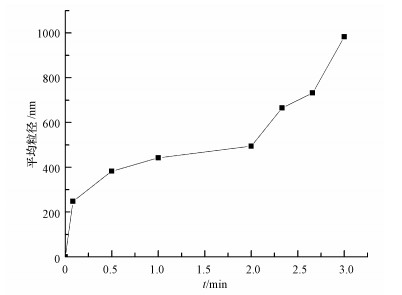
图 6 不同时间下单质硫颗粒粒度
由图 6可知, 当反应时间在0~5 s时, 体系中的单质硫颗粒粒径迅速增大至248.1 nm, 之后在5 s~2 min内其粒径随时间延长而缓慢增大, 在2~3 min内, 体系中单质硫颗粒粒径又由494.6 nm迅速增大至982.8 nm.出现这种现象的原因可能是在0~5 s时间段内, 主要是纳米晶体的形成阶段, 晶体形成的初始阶段是一个非常快速的过程;在5 s~2 min时间段内, 单质硫颗粒的增大主要是晶体的生长, 而纳米晶体生长的速度缓慢, 且受多种因素的限制(郝保红等, 2006).在2~3 min时间段内, 由于磁力搅拌器的搅拌作用及纳米硫颗粒本身的性质, 颗粒之间发生了团聚, 即多个纳米硫颗粒在某些因素的影响下聚集在一起, 形成了较大的颗粒.由单质硫颗粒的增大趋势可知, 单质硫颗粒迅速增大的阶段主要集中在2 min之后, 由此可以推测单质硫颗粒增大的主要原因是颗粒之间发生了团聚.
通过向反应体系加入分散剂聚丙烯酰胺(PAM), 考察单质硫颗粒增大的主要原因.在相同的反应条件下, 向1 L反应体系中分别加入10、20、30 mL质量分数为0.2%的PAM, 不同反应时间(1、3、5、7、9、11、13、15 min)下, 单质硫颗粒粒度如图 7所示.
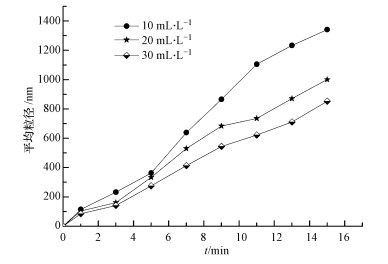
图 7 不同浓度PAM条件下单质硫颗粒粒度
由图 7可以看出, 向反应体系加入PAM后, 体系中的单质硫颗粒粒度随反应时间的延长而缓慢增大, 且在相同时间内随PAM浓度的增大而逐渐减小, 说明体系中加入的分散剂浓度越大, 单质硫颗粒的分散性越好.在0~15 min这段时间内, 体系中单质硫颗粒的粒度范围为85.2~1341.3 nm, 当反应时间为9 min时, 体系中粒度最大为865.1 nm;而相同条件下, 在未加分散剂的反应体系中, 当反应时间为3 min时, 单质硫颗粒粒度达到928.8 nm, 当反应时间为10 min时, 单质硫颗粒粒度已达到5.563μm.这说明分散剂的加入明显阻碍了单质硫颗粒的增大, PAM使单质硫分散的主要原因是其通过静电作用力或范德华力等在颗粒表面形成的吸附膜产生了强大的空间排斥效应(冯拉俊等, 2004), 这与Janssen等(1996)在研究微生物产生的硫颗粒的表面特性和机理时得出了相似的结论, 即链状高分子的存在会阻碍硫颗粒的聚合, 导致其呈现一种高度分散的状态, 而向体系中加入分散剂是控制颗粒团聚的有效方法之一.由此可知, 过氧化氢氧化含硫废水过程中生成的单质硫颗粒增大的主要原因是颗粒间的团聚.具体参见污水宝商城资料或http://www.dowater.com更多相关技术文档。
4 结论(Conclusions)
1) 通过控制反应体系氧化还原电位(ORP)可以控制氧化反应进程, 使单质硫成为主要氧化产物, 其收率随反应体系ORP值的升高先增大后减小, 在ORP为(30±5)mV时达到最大值76.35%;当ORP由(-50±5)mV升高至(50±5)mV时, 副产物S2O32-收率显著下降, 由26.54%降低至5.32%.
2) 通过XRD对反应过程中的固相产物进行晶体结构分析, 根据所得的X射线衍射图谱和标准图谱进行对比可知, 体系中所得的固相产物为单质硫, 且以结构稳定的环状分子S8、正交晶系斜方硫为主.
3) 通过SEM对反应过程中的单质硫进行观察可知, 液相中的单质硫是由多个极小的颗粒聚集而成, 并且随着时间的延长由最初的絮状变为薄层状, 然后逐渐向内部包裹最终形成大小不一、形状不规则的颗粒状单质硫; 结合Zeta电位及粒度分析可知, 随着反应时间的延长, 单质硫颗粒粒径由纳米级逐渐增大至微米级.
4) 通过对反应过程中单质硫颗粒粒度分析可知, 单质硫颗粒在0~5 s内迅速增大, 在5 s~2 min内增长缓慢, 在2~3 min时, 其粒径由494.6 nm迅速增大至982.8 nm, 通过向反应体系中加入分散剂证明了单质硫颗粒增大的主要原因是颗粒间发生了团聚.

