近半个多世纪以来, 全氟烷基酸类化合物(perfluoroalkyl acids, PFAAs)的广泛应用及其导致的环境污染, 引起了国内外学者的广泛关注. PFAAs被广泛用于金属电镀、农药、泡沫灭火剂、半导体、皮革、纺织、地毯、家具、纸制品、航空航天及食品容器等生产过程和产品中, 它的广泛使用导致其被直接或间接排放到环境中.目前在大气、水体、土壤、生物有机体组织及人体血液中都有PFAAs的检出. PFAAs在环境中具有高度稳定性, 它们可以进行生物富集, 并且存在着强大的碳-氟键使其具有潜在的生物毒性.因此, 在2009年5月召开的《关于持久性有机污染物的斯德哥尔摩公约》第四次缔约方大会上, 其家族成员全氟辛烷磺酸(perfluorooctane sulfonate, PFOS)及其盐和全氟辛基磺酰氟(perfluorooctane sulfonyl fluoride, POSF)被列入《斯德哥尔摩公约》, 将在全球范围内限制使用.所以, 有关全氟化合物的来源、分布、迁移转化等问题成为目前学术界研究的热点, 同时也引起了环境管理部门的高度重视.
此外, 众多研究已经证实, PFAAs的前体物与其中间产物在环境中也广泛存在, 如全氟辛烷磺酰胺(perfluorooctane sulfoneamide, FOSA)、8:2氟调聚醇(8:2 fluorotelomer alcohols, FTOHs)就是环境介质中常被检出的前体物, 这些前体物最终转化成PFAAs, 如FOSAs在好氧条件下可以转化生成PFOS, FTOHs的降解可以生成全氟辛酸(perfluorooctanoic acid, PFOA), FTOHs在大气环境中也可以转化为全氟羧酸(perfluorinated carboxylic acids, PFCAs), 所以, 前体物和中间产物也是PFAAs在环境中的重要潜在来源.然而, 由于相应分析测试技术和仪器条件的缺乏, 到目前为止针对环境介质中全氟化合物前体物产生和转化的系统研究仍然很少, 仅有少数前体物或中间产物被测得; 绝大多数化合物还不能被直接检测和分析, 还处于方法的探索阶段; 如运用羟基自由基氧化法, 通过对比氧化前后溶液中一定碳链长度化合物的浓度差异, 对前体物和中间产物的含量水平进行计算的方法证实了前体物和中间产物是水环境中PFAAs的重要潜在来源.然而, 目前我国在该方面开展的研究还很缺乏, 特别是针对城市不同水体中PFAAs前体物转化潜力及对环境污染贡献的研究几乎还是空白.
日照市是山东省东南部的重要沿海开放城市, 也是典型旅游型城市.近些年来, 旅游业成为该市的主导产业, 同时钢铁、纸浆和造纸、纺织等工业也发展迅速.环境监测表明日照市整体环境质量良好; 但随着该市工农业的快速发展, 日益严峻的环境污染形势不容乐观.如何在大力发展旅游业, 保持良好生态环境质量的同时保持社会经济的快速健康发展, 是日照市近年来积极探索的发展道路.然而到目前为止, 有关日照市环境污染状况的研究相对较少, 对于该市各类水体中新型污染物研究的报道更是空白.
因此, 本文以日照市主要河流水体与 污水厂出水为研究对象, 对这些水体中PFAAs的含量与空间分布及其前体物转化对PFAAs污染的贡献进行系统探讨, 以期为日照市地表水体及其附近海域环境保护提供基础数据, 也为我国旅游型城市新型污染物环境污染的有效防治提供数据支撑和借鉴.
1 材料与方法 1.1 水样的采集
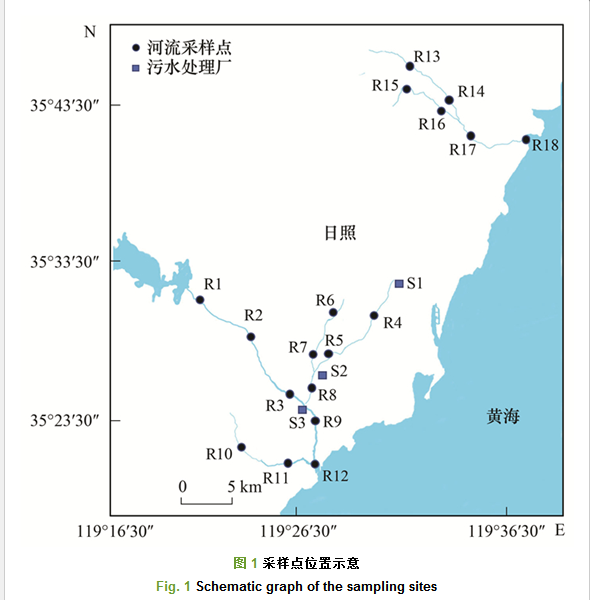
2017年5月, 在日照市付疃河、潮河与市区重要排水沟渠(R1~R18)及市内3座污水厂(S1~S3)出水口采集水体样品, 采样点位置如图 1所示.用5 L的有机玻璃采水器采集河水0~20 cm的表层水, 在出水口采集污水厂出水样品.用Whatman玻璃纤维滤膜(1.2 μm)对采集的所有水样进行过滤, 目的是去除水体中的悬浮颗粒物, 过滤的水样装入事先用去离子水和甲醇润洗过的高密度聚乙烯(HDPE)瓶中.采样过程中用超纯水做样品空白, 与所采集的水样做同步预处理.为了防止PFAAs污染, 在采样、预处理及实验全过程中严禁接触和使用含聚氟的任何材料和实验器皿.
试剂和仪器:本文所使用的仪器包括超高效液相色谱质谱联用仪(UPLC-MS/MS, Waters Acquity UPLC-Quattro Premier XE型), 弱阴离子交换柱(WAX柱, Qasis® WAX, 6 cc, 150 mg, 30 μm), Waters BEH-C18的色谱柱(2.1 mm×50 mm, 1.7 μm), Agilent聚丙烯液相小瓶(1mL), 氮吹仪(Organomation Associates公司, 加拿大), 玻璃纤维滤膜(1.2 μm, Whatman公司), Amberlite XAD-2树脂(Supleco, 美国), Milli-QA10去离子水发生器(美国Millipore公司).
本文所使用的试剂包括:全氟丁酸(perfluorobutyric acid, PFBA)、全氟戊酸(perfluoropentanoic acid, PFPA)、全氟庚酸(perfluoroheptanoic acid, PFHpA)、全氟己酸(perfluorohexanoic acid, PFHxA)、全氟癸酸(perfluorodecanoic acid, PFDA)、全氟辛酸(perfluorooctanoic acid, PFOA)、全氟壬酸(perfluorononanoic acid, PFNA)、全氟十一酸(perfluoroundecanoic acid, PFUnDA)、全氟十二酸(perfluorododecanoic acid, PFDoDA)、全氟十三酸(perfluorotridecanoic acid, PFTrDA)、全氟十四酸(perfluorotetradecanoic acid, PFTeDA)、全氟癸烷磺酸(perfluorodecanesulfonic acid, PFDS)、全氟丁烷磺酸(perfluorobutanesulphonic acid, PFBS)、全氟辛烷磺酸(perfluorooctanesulfonic acid, PFOS)、全氟己烷磺酸(perfluorohexanesulfonic acid, PFHxS)、全氟辛基磺酰胺(perfluorooctane sulfoneamide, FOSA)、8:2调聚全氟辛基羧酸酯(2-perfluorooctylethanoate, 8:2FTCA)、全氟辛基磺胺乙醇磷酸酯(perfluorooctane sulfonamidoethanol-based phosphate diester, di-SAmPAP), 内标物[13C4]PFBA、[13C4]PFOS、[13C4]PFOA、[13C5]PFNA、[13C2]PFHxA、[13C2]PFDA、[13C2]PFUnDA、[13C2]PFDoDA、[18O2]PFHxS, 上述试剂全部购自加拿大Wellington公司, 纯度均大于98%;甲醇(美国Tedia公司, 为色谱纯级); 氨水(25%, 比利时Acros公司)为优级纯.
超高效液相色谱的测试条件:Waters BEH-C18的色谱柱(2.1 mm×50 mm, 1.7 μm); 流动相A为含2 mmol乙酸铵的水和甲醇混合液(98:2);流动相B为含2 mmol乙酸铵的甲醇溶液; 色谱柱的温度为40℃, 流速为0.30 mL・min-1; 进样量为5 μL.
质谱条件:电喷雾离子源, 负离子扫描(ESI-), 多反应监测(MRM)模式, 碰撞气(Ar)流速0.18 mL・min-1, 辅助气(N2)流速10 mL・min-1, 雾化温度380℃, 离子源温度120℃.
1.3 PFAA前体物氧化处理
本文采用羟基自由基(・OH)氧化法分析水样中PFAAs前体物的含量及转化潜力.方法简述如下:在碱性条件下利用过硫酸盐(S2O82-)热解产生羟基自由基, 利用该羟基自由基的强氧化能力将PFAAs前体物进行氧化转化; 对样品进行氧化处理后, 对氧化前后溶液中PFAAs的浓度进行比较, 确定前体物的含量水平.具体步骤为:将过硫酸盐(2 mL, 60 mmol・L-1)和氢氧化钠(1.9 mL, 150 mmol・L-1)加入到装有未过滤水样的HDPE瓶(125 mL)中, 每个样品做3个重复, 将HDPE瓶置于油浴锅中85℃加热6 h, 冷却至室温.
1.4 水样前处理
本文所运用的提取程序如下:依次用4 mL氨水(0.1%)的甲醇溶液、4 mL甲醇、4 mL水进行活化, 然后向1.0 L已经过滤的水样中加入10 ng相应内标并摇匀, 将上述加内标的水样以1.0 mL・min-1的速度流过WAX小柱, 使待测污染物吸附在WAX小柱上.过滤完毕后, 用4 mL浓度为25 mmol・L-1的醋酸盐缓冲液(pH=4)冲洗WAX小柱, 然后将该小柱离心除去残留的水, 用4 mL甲醇和含4 mL 0.1%氨水的甲醇溶液对目标化合物进行洗脱, 洗脱液经氮吹后, 用初始流动相定容至1.0 mL, 最后, 采用UPLC-MS/MS测定15种全氟酸类化合物和3种前体物质的质量浓度.
1.5 质量保证与控制
实验以超纯水进行全过程空白对照, 实验所用器皿均为聚丙烯材质, 所有实验器具使用前均用甲醇和超纯水进行洗涤.在1 L空白水样中加入所有待测物的标准溶液(含10 ng)混标和内标物质(10 ng), 按照与样品相同的实验条件进行回收率分析测试.分析结果显示:水样中各待测物的加标回收率为79.95%~110.36%, 相对标准偏差范围为3.37%~7.12%.水样中的PFASs的检测限为0.06~0.18 ng・L-1, 空白对照样品中未检出PFAAs.
1.6 数据统计
本文应用SPASS 13.0、EXCEL 2016等软件对实验数据进行分析.
2 结果与讨论 2.1 氧化处理前河流水体和污水厂出水中PFAAs及其前体物的含量和空间分布
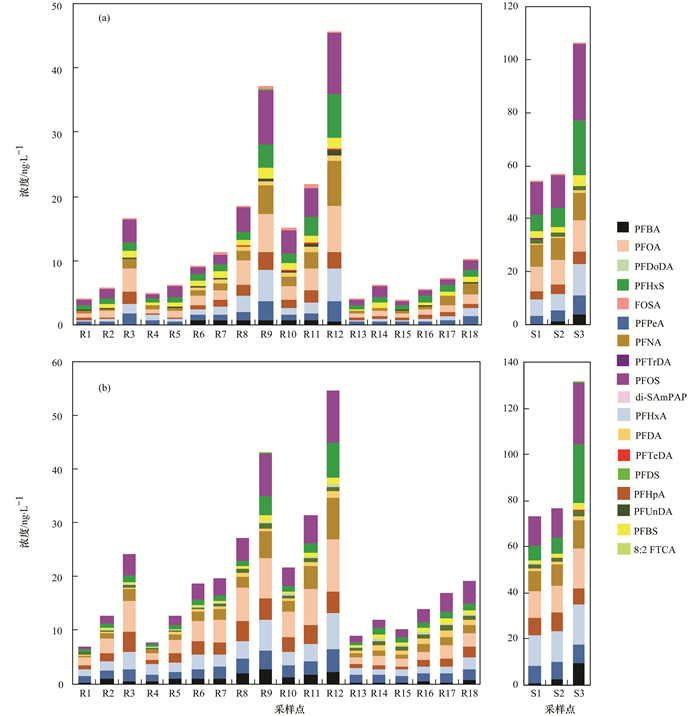
对日照市河流水体与污水厂出水中15种PFAAs(包括碳原子数4~14的全氟烷基羧酸类化合物(PFCAC4~C14)和碳原子数为4、6、8、10的全氟烷基磺酸类化合物(PFSAC4, 6, 8, 10)及3种PFAAs前体物(8:2 FTCAs, FOSA, di-SAmPAP)的质量浓度进行分析, 结果如图 2所示.结果表明:15种PFAAs在绝大多数水样中均被检出, 所以PFAAs在日照市河流和污水厂出水中广泛分布.在所有样品中, 15种PFAAs及其3种前体物的总质量浓度范围为3.93~106.18 ng・L-1(表 1), 其中PFOS、PFOA、PFHxS和PFNA是质量浓度较高的4种物质, 质量浓度范围分别为0.66~28.76、0.68~11.68、0.57~20.54和0.35~10.34 ng・L-1, 其均值分别为4.77、3.26、2.94和2.55 ng・L-1, 所以PFOS和PFOA是质量浓度最高的两种污染物; 在绝大多数样品中PFBA、PFDoDA、PFTrDA和PFDS的质量浓度较低或不能被检出; 而在所有样品中, 3种前体物的质量浓度均低于PFCAs和PFSAs, 只有FOSA检出率较高(表 1).
|
表 1 氧化处理前河流与污水处理厂废水水样中PFAAs及其前体物的质量浓度/ng・L-1
|
表 1 氧化处理前河流与污水处理厂废水水样中PFAAs及其前体物的质量浓度/ng・L-1 Table 1 Concentrations of PFAAs and their precursors in water samples of the rivers and sewage treatment plants effluents before oxidation treatment/ng・L-1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

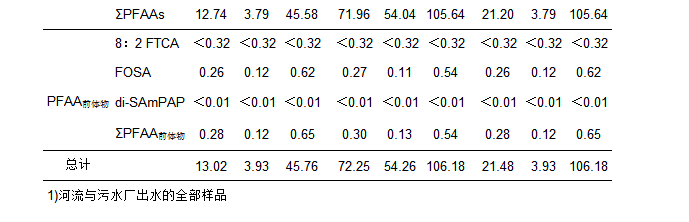
河流水样中, 氧化前15种PFAAs及其3种前体物的总质量浓度范围为3.93~45.70 ng・L-1, 均值为13.02 ng・L-1; 其中质量浓度较高的为PFOS、PFOA、PFHxS和PFNA, 其值分别为0.66~9.65、0.68~7.31、0.57~6.79和0.35~6.94 ng・L-1; 其均值分别为2.59、2.13、1.56和1.49 ng・L-1(表 1).河流18个采样点的总浓度具有明显的空间差异, 每一条河流下游浓度明显高于上游(图 2), 其原因可能是河流上游的采样点主要位于农村地区, 周围没有工业污染物的排放和影响, 所以PFAAs的浓度较低; 如R1点为城市饮用水源地, 没有任何污染物排放, 所以其浓度很低; 河流下游采样点位于城区和工业区附近, 受周围工业废水和生活污水排放的影响, 所以PFAAs的浓度较高.另外, 本文选取的三座污水处理厂, 其出水全部排入到附近河道中, 对河流污染物的含量可能有重要影响; 如R12点就是各项来水的汇集点, 该点ΣPFAAs值最高.所以, 河流上下游接纳污染物数量的不同可能是导致河流不同采样点PFAAs浓度空间差异的重要原因.此外, 人口密度、区域工业发展程度及居民生活水平也可能影响附近水环境中全氟化合物的浓度[6, 20].
在污水厂出水中, 15种PFAAs及其3种前体物的总质量浓度为54.26~106.18 ng・L-1, 均值为72.25 ng・L-1(表 1), 其中质量浓度最高的几种PFAAs为PFOS(12.28~28.76 ng・L-1, 平均为17.82 ng・L-1)、PFHxS(6.42~20.54 ng・L-1, 平均为11.26 ng・L-1)、PFOA(9.18~11.68 ng・L-1, 平均为10.06 ng・L-1)、PFNA(8.12~10.34 ng・L-1, 平均为8.93 ng・L-1), 见表 1.因此, 污水厂出水中PFAAs的质量浓度明显高于河流水体.以前的研究结果表明:沈阳、南京、大连和广州的污水厂出水ΣPFAAs及其前体物的质量浓度水平与本文结果相似[21].在本文中所调查的3座污水厂出水都注入附近河流并最终汇入黄海, 因此, 污水厂排放是河流水体不可忽视的污染源.
对河流和污水厂出水中8:2 FTCA、FOSA、di-SAmPAP这3种物质进行分析的结果表明:3种前体物的质量浓度都远低于PFCAs和PFSAs. 3种前体物中, 只有FOSA被检出, 而8:2 FTCA和di-SAmPAP在所有采样点中都未被检出; 其中, 河流水体中FOSA的质量浓度范围为0.12~0.62 ng・L-1, 均值为0.26 ng・L-1; 污水厂出水中FOSA的质量浓度范围为0.11~0.54 ng・L-1, 均值为0.27 ng・L-1; 所以, 污水厂出水中FOSA的质量浓度值与河流水体相当.
2.2 氧化处理后河流水体和污水厂出水中PFAAs及其前体物的含量及其变化
对河流水体和污水厂出水所有样品进行羟基自由基氧化处理后发现, PFCAs的质量浓度有了显著升高, 而PFSAs的质量浓度没有明显变化(图 3).此结果表明, 羟基自由基氧化处理的方法可以将PFAAs的前体物氧化转化成PFCAs, 同时也证明前体物是水环境中PFAAs的潜在源.氧化处理后水体中PFCAs的总含量也就包括氧化前已存在的(PFCAs氧化前)和前体物氧化转化生成的(ΔPFCAs)两个部分之和.
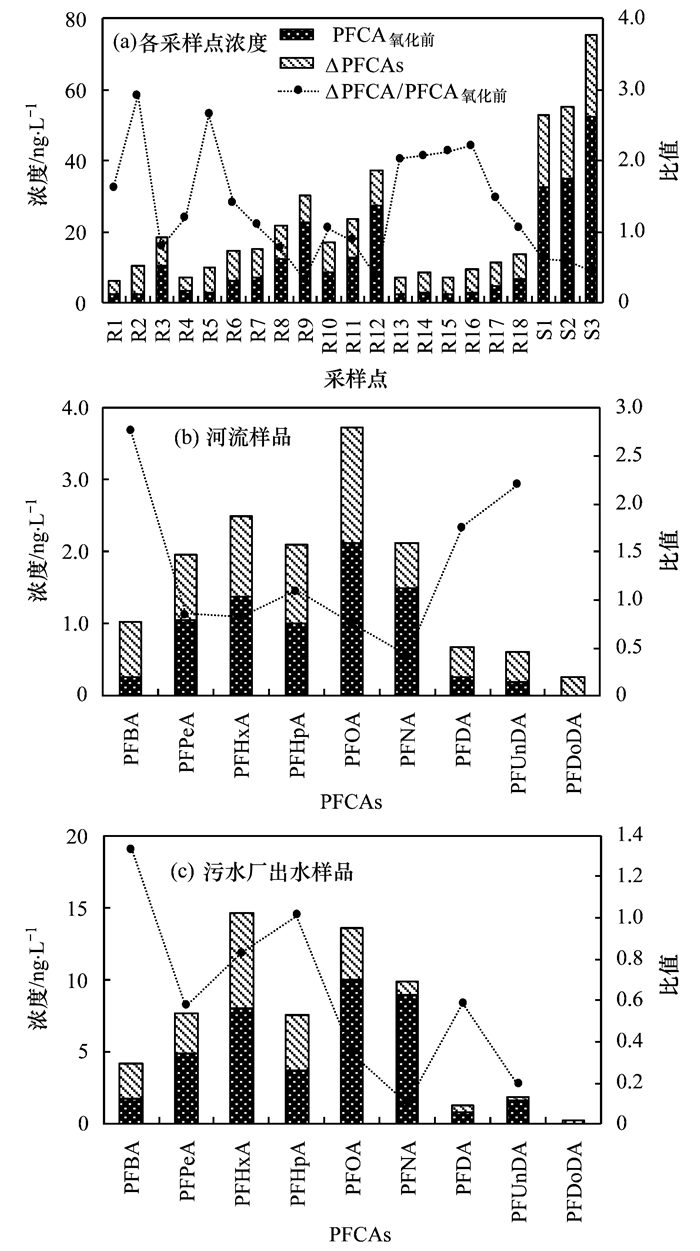
河流水体中, 碳原子数为4~12的PFCAs(PFCAC4~C12)氧化后质量浓度增加值(ΣΔPFCAC4~C12)的范围为3.82~11.08 ng・L-1, 平均为7.22 ng・L-1; 污水厂出水中ΣΔPFCAC4~C12的值20.13~23.21 ng・L-1, 均值为21.22 ng・L-1 (表 2); 其中, 在河流水体中浓度变化较大的PFCAs分别是PFOA(0.55~3.20 ng・L-1, 均值为1.61 ng・L-1)、PFHxA(0.63~1.86 ng・L-1, 均值为1.12 ng・L-1)、PFHpA(0.25~2.07 ng・L-1, 均值为1.10 ng・L-1).在污水厂出水中质量浓度增加较大的PFCAs分别是PFHxA(6.07~7.05 ng・L-1, 平均为6.65 ng・L-1)、PFHpA(2.35~4.6 ng・L-1, 平均3.82 ng・L-1)、PFOA(2.38~5.63 ng・L-1, 平均为3.49 ng・L-1).因此, 在氧化前后PFAAs及其某些同系物的质量浓度变化明显, 从而说明PFAAs前体物在水环境中的存在具体联系污水宝或参见http://www.dowater.com更多相关技术文档。 .
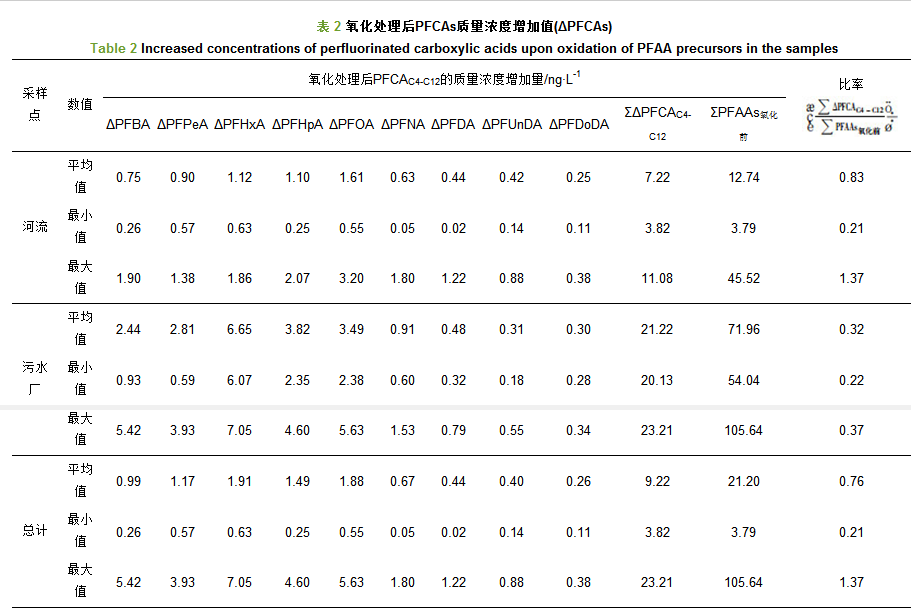
从以上结果也可发现, 在河流和污水厂出水所有样品中, 浓度升高最多的是碳原子数为4~8的PFCAs化合物(PFCAC4~C8), 这些化合物浓度的增加值远大于碳原子数在9~12的PFCAs化合物(PFCAC9~C12), 这可能由于在氧化过程中, 相比于长链化合物, 更多的前体物被转化成短链PFCAs.然而, PFSAs的质量浓度在氧化前后并没有明显差异; 因此, 氧化后PFAAs前体物不能转化成PFSAs化合物.因此, PFAAs前体物对水环境的影响不可忽视.
此外, 在绝大多数样品中, 碳原子数多于12的PFCAs化合物(PFTrDA和PFTeDA)的质量浓度在氧化后略有下降, 这与以前的研究结果一致[18].有研究已证实, 在基于羟基自由基氧化过程中, PFAAs前体物的转化与全氟烷基碳链长度有关[19, 22, 23].在碱性环境下, 含n个碳原子的含磺酰胺的前体物可转化为碳原子数相同的PFCAs, 含n个碳原子的氟调聚物的前体物可转化为含4~(n+1)个碳原子的PFCAs[22, 23].本文的研究结果还可以看出, 在河流的不同位置, 氧化后PFCAC4-C12的质量浓度增加量(ΣΔPFCAC4~C12)有明显不同, 表现出明显的空间差异性(图 3); 同时, 同一条河流下游的ΣΔPFCAC4~C12明显高于上游; 例如本文涉及的付疃河位于日照市的中部, 是日照市最大的河流, 3个污水处理厂出水最终全部汇入该河流, 其慢速流动的性质可能有利于前体物的迁移转化与降解; 因此, 所有这些因素可能是导致该河流中氧化后ΣΔPFCAC4~C12较高的重要原因. ΣΔPFCAC4~C12的最大值和最小值分布在河流的下游和上游, 此结果反映了PFAA前体物含量沿河流自上游向下游逐渐增加的趋势.相比于河流水体, 氧化后污水厂出水ΣΔPFCAC4~C12的值更高(图 3), 此结果表明污水厂排水对河流水环境PFAAs污染可能会有所贡献.
本文通过对PFCAs质量浓度的增加量(ΔPFCAs)与氧化前PFCAs化合物(PFCA氧化前)的质量浓度之比(ΔPFCAs/PFCA氧化前)进行了分析, 揭示了PFAAs前体物的转化特征(图 3和表 3).在河流水体样品中, 比值较高化合物为PFUnDA(0.00~8.50, 平均为2.78)、PFHxA(0.13~3.36, 平均为1.51)、PFHpA(0.51~4.09, 平均为1.49);在污水厂出水中, 比值较高的化合物为PFHpA(0.51~1.44, 平均为1.08)、PFHpA(0.51~1.14, 平均为0.93)、PFPeA(0.08~1.08, 平均为0.72);所以, 此比值在不同水体中存在明显差异.此外, 污水厂出水的ΔPFCAs要远高于在河流水体, 而污水厂出水ΔPFCAs/PFCA氧化前值却明显低于河流水样的值.污水厂出水中ΔPFCAs较高, 而比值较低, 这可能由于污水处理过程中前体物的降解所致.
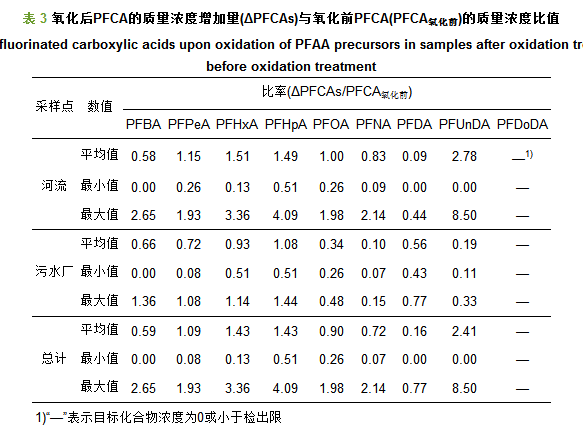
总之, 在氧化处理前后, PFAAs含量的变化揭示了水体中PFAAs前体物的存在; 氧化后PFCAs浓度的增加主要是由PFAAs前体物的转化所致.本文只选取了3种典型的前体物进行探索, 在实际水环境中肯定还存在其它PFAAs前体物, 其转化机制还有待于进一步研究.
2.3 PFAAs的来源分析
根据已有的研究结果, 基于全氟化合物同系物浓度的比值, 可以考察PFAAs的可能潜在来源, 如PFOS/PFOA、PFHpA/PFOA、PFOA/PFNA等之间的比值可以作为参考; 如PFOS/PFOA大于1.0时, 则可能存在PFAAs的点源污染.本文涉及的日照城市水体中PFOS/PFOA的范围为0.99~1.75, 高于朝鲜沿海水域、太湖、长江、南四湖、密西西比河下游河段[30]的相应值, 这说明了日照市河流附近区域可能存在点源污染.
本文中PFOA/PFNA的值在1.32~2.75之间, 根据已有的研究结果, 当PFOA/PFNA的值在7~15之间时, 水环境可能受氟化制造业直接排放的影响[31]; 当PFOA/PFNA的值在1.7~56.8之间时, 水体可能受到PFAAs二次来源的影响, 如挥发性前体物降解[32]; 而当PFOA/PFNA的值小于1.5时, 可能是仅接收挥发性前体物大气沉降的偏远地区[33].根据以上结论, 说明日照市水体环境可能存在PFAAs的二次污染源, 如挥发性前体物降解, 影响了日照市水体中全氟化合物的含量.所以, PFAAs前体物的降解转化是日照市河流水体PFAAs的一个重要来源.有关具体的源解析, 需要在以后的研究中进一步探索.
3 结论
(1) 日照市河流水体和污水厂出水15种PFAAs及其3种前体物的总质量浓度范围为3.93~45.70 ng・L-1和54.26~106.18 ng・L-1; 河流水体和污水厂出水中含量占绝对优势的污染物为PFOS、PFOA、PFHxS和PFNA; 污水厂出水中PFAAs的平均质量浓度明显高于河流水体, 污水厂出水对河流水体PFAAs污染可能有所贡献.
(2) 空间分布上, 不同河流和污水厂出水中PFAAs质量浓度差异明显; 河流上游PFAAs质量浓度明显低于下游; 城市生活污水和工业废水排放及污水厂出水等的汇入对河流水体PFAAs的污染有重要影响.
(3) 运用羟基自由基氧化处理后, 日照市河流水体和污水厂出水中PFCAs的质量浓度显著提高, 其中以PFCAC4~C12升高尤为明显, 而PFSAs的质量浓度没有明显变化; 其中河流水体和污水厂出水中ΣΔPFCAC4~C12的范围为3.82~11.08 ng・L-1和20.13~23.21 ng・L-1; 河流水体ΣΔPFCAC4~C12的平均值明显低于污水厂出水; 河流水体中ΣΔPFCAC4~C12下游明显高于上游, 不同河流ΣΔPFCAC4~C12也存在明显差异; 所以, 氧化处理前后PFAAs浓度的明显差异, 验证了水体环境中PFAAs前体物的存在; 污水厂出水可能是河流水体中PFAAs前体物的重要来源.
(4)氧化处理后PFCAC4~C8的质量浓度增加量远大于PFCAC9~C12, 氧化处理导致更多的前体物转化成短链PFCAs; 污水厂出水中前体物的转化率低于河流水体, 所以污水处理过程中可能存在前体物的降解.(来源:环境科学 作者:王世亮, 孙建树, 杨月伟, 张敏)

