1 引言
煤矿或各种有色金属矿在开采与废矿石堆放过程中,常使与矿层伴生的硫铁矿暴露于空气中与 地下水或地表水中,通过系列化学与生物氧化过程,使得近中性的地下水转变为低pH、高Fe、SO2-4,且多种重(类)金属离子(Cd、Pb、Cu、Zn、As等)并存的酸性矿山废水(acid mine drainage,AMD).此类废水若不经有效处理而任意排放,将严重污染地表水及土地资源,威胁农作物、水生生物与人体健康.
石灰中和法是世界上最常用的AMD治理方法.然而,大多数AMD体系中含有较大量的Fe2+,由于Fe(OH)2 离子浓度积(1.6×10-14,18 ℃)远大于Fe(OH)3的离子浓度积(1.1×10-36,18 ℃),所以为了在近中性条件下使得Fe离子完全沉淀,在工程应用中,常常在化学中和前段完成Fe2+氧化过程.以AMD为介质,利用氧化亚铁硫杆菌(A. ferrooxidans)生物氧化Fe2+进而合成次生铁矿物(施氏矿物、黄铁矾类物质)不仅可以有效去除AMD中存在一定量的Fe与SO2-4,且此类次生铁矿物在合成过程中亦可通过吸附与共沉淀方式大幅度去除体系中的Cu、Cd、Hg、Pb、As等有毒有害元素.另需要强调的是对于石灰中和法得到的Fe(OH)3絮状凝胶而言,施氏矿物与黄铁矾类物质沉降性能良好,易于沉淀,可以极大降低后续固液分离成本.因此,前期氧化亚铁硫杆菌(A. ferrooxidans)生物氧化Fe2+产生次生铁矿物与后期化学中和相结合的工艺在AMD的治理领域表现出一定的应用潜力.
由于煤矿及其它有色金属矿中常有含镁矿物(白云石富镁碳酸盐矿物、蛇纹石与绿泥石等富镁硅酸盐矿物等)的存在,使得产生的AMD中含有一定量的Mg2+.研究证实,A. ferrooxidans菌体及其胞外多聚物可以作为次生铁矿物合成的晶种.而Mg2+可以在微生物胞外多聚物之间形成架桥使得微生物菌体团聚.那么,这一团聚过程是否会使得矿物较易在反应器壁粘附,进而影响次生铁矿物合成体系总Fe沉淀率及矿物的形貌?另外,高的转速对应高的剪切力.那么,高转速是否会减缓矿物在反应器壁的粘附行为?为了探究此类科研问题,本研究分别在不同培养转速条件下,考察了Mg2+浓度不同对A.ferrooxidans催化合成次生铁矿物体系Fe2+氧化率、总Fe沉淀率、次生铁矿物反应器壁粘附状况及矿物形貌的影响.以期为生物合成次生铁矿物工艺的优化及其在酸性矿山废水治理领域的成功应用提供一些必要的参数.
2 材料与方法
2.1 嗜酸性氧化亚铁硫杆菌(A. ferrooxidans)接种液的制备
在150 mL改进型9K液态培养基(FeSO4 ・ 7H2O 44.24 g、(NH4)2SO4 3.0 g、KCl 0.10 g、K2HPO4 0.50 g、Ca(NO3)2 ・ 4H2O 0.01 g、MgSO4 ・ 7H2O 0.50 g,去离子水1 L)中接种A. ferrooxidans LX5(CGMCC No.0727),体系用H2SO4调节pH至2.5后,置于180 r ・ min-1往复式振荡器(ZD-85A恒温振荡器)中在28 ℃培养2~3 d至体系Fe2+完全氧化.培养液经定性滤纸过滤以除去沉淀,过滤所得的液体即为嗜酸性氧化亚铁硫杆菌菌液.将所得菌液15 mL接种于135 mL改进型9K液态培养基中重复上述过程.所获菌液即为本研究后续次生铁矿物合成所需的微生物接种菌液,菌密度约为107 cells ・ mL-1.
2.2 生物合成次生铁矿物试验
在一系列250 mL锥形瓶中分别盛放制备好的A. ferrooxidans LX5接种液15 mL,①加入浓缩10倍的改进型9K液体培养基(Mg2+浓度为480 mg ・ L-1,以MgSO4 ・ 7H2O形式加入)15 mL,后补充去离子水至溶液总体积为150 mL,使得体系Mg2+浓度为48 mg ・ L-1(记作“Mg2+-48 mg ・ L-1”体系);②其它试验设计同处理①,而体系Mg2+设计浓度为4.8 mg ・ L-1(记作“Mg2+-4.8 mg ・ L-1”体系).用H2SO4将上述pH调至2.50,分别将混合液在28 ℃,180 r ・ min-1或100 r ・ min-1条件下振荡培养,每个处理设置3个重复.每12 h监测体系pH值,且从体系均匀取样1 mL,过0.45 μm滤膜,测定滤液Fe2+及总Fe浓度,进而计算Fe2+氧化率及总Fe沉淀率.待体系Fe2+氧化完全后将不同体系产生的矿物沉淀用定性滤纸收集,酸化的去离子水(pH=2.0)洗3次,再用去离子水洗涤2次后,在50 ℃环境中烘干,分析矿物的矿相及形貌.
2.3 测定方法
溶液pH用PHS-3C型酸度计测定,Fe2+与总Fe浓度采用邻菲罗啉比色法进行分析. t时刻Fe2+氧化率=(C (Fe2+)0-C (Fe2+)t)/ C (Fe2+)0×100%,式中,C (Fe2+)0与C (Fe2+)t分别为反应初始和反应t小时体系Fe2+浓度.t时刻总Fe沉淀率=(C (TFe)0-C (TFe)t)/ C (TFe)0×100%,式中C (TFe)0与C (TFe)t分别为反应初始和反应t小时体系总Fe浓度.次生铁矿物矿相用X射线衍射仪(XRD,MiniFles II,日本理学)测定,测试工作条件为:管电压30 kV,管电流15 mA,扫描区间10~70°(2θ),步长0.02°,Cu靶(弯晶单色器).次生铁矿物形貌采用热场发射扫描电子显微镜(SEM,JSM-7001F)观察,工作距离(样品表面到物镜的距离)9.7 mm,加速电压5.0 kV.
3 结果与分析
3.1 不同培养转速下Mg2+对生物合成次生铁矿物体系pH的影响
Fe2+生物氧化为Fe3+是一个能使体系pH升高的过程,后续Fe3+水解产生次生铁矿物却是pH降低的过程.180 r ・ min-1或100 r ・ min-1的培养转速下,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两生物合成次生铁矿物体系pH随时间的变化情况如图 1所示.可以得出,转速对体系Fe2+生物氧化至Fe3+及后续Fe3+水解过程有明显的影响.当培养转速为180 r ・ min-1时,“Mg2+-4.8 mg ・ L-1”生物合成次生铁矿物体系pH首先从0 h的~2.50升高至12 h的 ~2.65,后逐渐降低至48 h的~2.07.“Mg2+-48 mg ・ L-1”体系在此培养转速条件下,pH首先从0 h的~2.50升高至12 h的 ~2.66,后逐渐降低至48 h的~2.12.然而,当培养转速为100 r ・ min-1时,体系pH下降速度相对缓慢,“Mg2+-4.8 mg ・ L-1”体系与“Mg2+-48 mg ・ L-1”体系pH在12 h均达到~2.69,后逐渐分别降低至72 h的~2.21与~2.17.
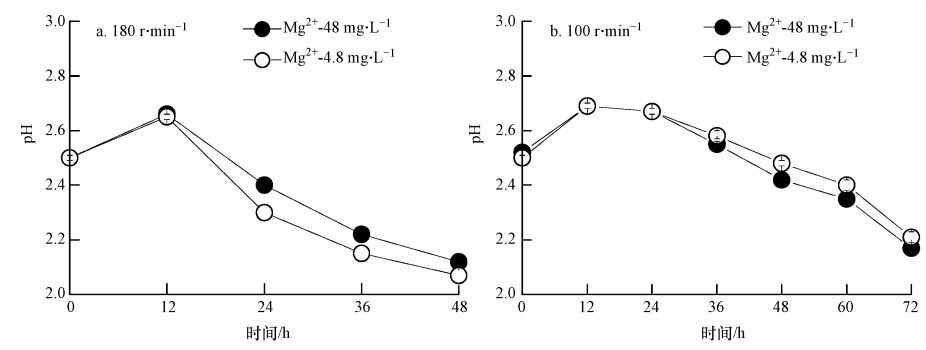
图 1 A. ferrooxidans催化合成次生铁矿物体系不同培养转速条件下pH变化情况
整体而言,不同培养转速下,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”体系pH变化差异主要表现在pH下降阶段.然而,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两体系pH降低快慢趋势却随培养转速的不同而不尽相同.在培养转速为180 r ・ min-1条件下,“Mg2+-4.8 mg ・ L-1”体系pH下降速度要快于“Mg2+-48 mg ・ L-1”体系,而当培养转速为100 r ・ min-1 时,后者pH下降速度却快于前者.
3.2 不同培养转速下Mg2+对生物合成次生铁矿物体系Fe2+氧化率的影响
体系Fe2+有效氧化是后续Fe3+水解产生次生铁矿物的前提条件.本研究中,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两生物合成次生铁矿物体系在180 r ・ min-1或100 r ・ min-1的培养转速下,Fe2+氧化率随时间的变化趋势见图 2.
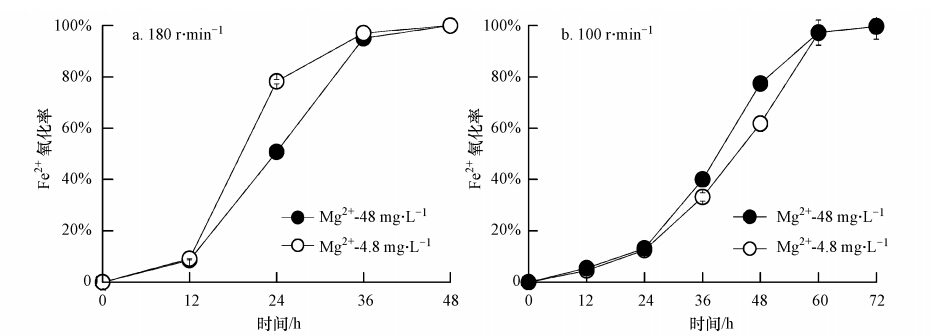
图 2 A. ferrooxidans催化合成次生铁矿物体系不同培养转速条件下Fe2+氧化率变化趋势
从图 2可以得出,当培养转速为180 r ・ min-1 时,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两体系均可在48 h内实现Fe2+完全氧化.而当转速降低至100 r ・ min-1 时,两体系Fe2+完全氧化时间要延长至72 h.然而,任一体系次生铁矿物合成过程中,Fe2+氧化率随培养时间均呈现“S型”变化趋势.例如,“Mg2+-4.8 mg ・ L-1”体系在180 r ・ min-1的培养条件下,Fe2+氧化率在前12 h仅为9.1%,12~36 h 培养过程中,Fe2+氧化率聚增至97%,体系培养至48 h时,Fe2+氧化完全.同时可以发现,当培养转速为180 r ・ min-1时,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两体系Fe2+氧化率在0~12 h与36~48 h“首尾”培养期间并不存在明显差异.而在12~36 h 过程中,“Mg2+-4.8 mg ・ L-1”体系Fe2+氧化率要明显快于“Mg2+-48 mg ・ L-1”体系.例如,在培养至24 h时,“Mg2+-4.8 mg ・ L-1”体系Fe2+氧化率达到78.2%,而“Mg2+-48 mg ・ L-1”体系仅为50.8%.同样,当培养转速为100 r ・ min-1时,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两体系Fe2+氧化率在0~24 h与60~72 h“首尾”期间亦不存在明显差异.差异主要集中表现在24~60 h的培养过程中,然而,与180 r ・ min-1培养条件所得结果不同,此时“Mg2+-4.8 mg ・ L-1”体系Fe2+氧化率却稍慢于“Mg2+-48 mg ・ L-1”体系.例如,在36 h与48 h 时,“Mg2+-4.8 mg ・ L-1”体系Fe2+氧化率分别为33.2%与61.9%,而相应的“Mg2+-4.8 mg ・ L-1”体系Fe2+氧化率却分别为40.0%与77.3%.
3.3 不同培养转速下Mg2+对生物合成次生铁矿物体系总Fe沉淀率的影响
一般而言,次生铁矿物合成体系总Fe沉淀率越高,意味着较多的Fe参与体系次生铁矿物的合成.所以,总Fe沉淀率变化情况能够直接反应体系次生铁矿物合成能力.此研究中,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两生物合成次生铁矿物体系在180 r ・ min-1或100 r ・ min-1培养转速下,总Fe沉淀率随时间变化趋势如图 3所示.
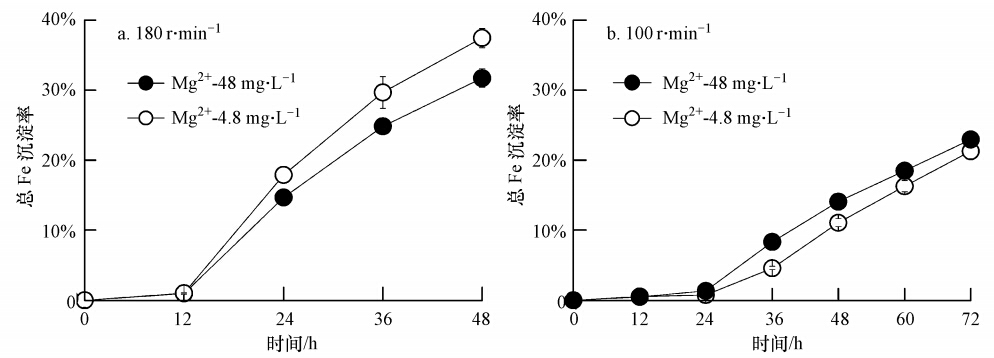
图 3 A. ferrooxidans催化合成次生铁矿物体系不同培养转速条件下总Fe沉淀率变化趋势
从图 3可以得出,当培养转速为180 r ・ min-1 时,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两体系培养前12 h总Fe沉淀率几乎可以忽略,而在后期培养过程中,两体系总Fe沉淀率均呈现逐渐增加趋势,然而增加的幅度却不尽一致.“Mg2+-4.8 mg ・ L-1”体系在12~48 h培养过程中,总Fe沉淀率从1.0%增加至37.4%.而“Mg2+-4.8 mg ・ L-1”体系在相应时间,总Fe沉淀率仅从1.0%增加至31.7%.前者总Fe沉淀率较后者提高近18.0%.而当培养转速为100 r ・ min-1 时,上述两矿物合成体系在培养前24 h总Fe沉淀率亦可以忽略,培养后期总铁沉淀率逐渐增加.培养至72 h 体系Fe2+完全氧化时刻,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两体系总Fe沉淀率分别为21.3%与23.0%,前者较后者降低7.4%.需要说明的是,相同矿物合成体系,高转速培养更利于总Fe沉淀转化为次生铁矿物.图 3结果表明,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两体系在180 r ・ min-1条件下培养至Fe2+完全氧化时,体系总Fe沉淀率较同体系在100 r ・ min-1培养条件下所得总Fe沉淀率增加了75.6%与37.8%.
3.4 不同培养转速下Mg2+对生物合成次生铁矿物矿相的影响
X射线衍射图谱(XRD)技术常常被用来分析矿物的矿相.本研究“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”体系在不同转速下培养,当Fe2+完全氧化时的矿物X射线衍射图谱见图 4.

图 4 生物合成次生铁矿物体系亚铁完全氧化时矿物X射线衍射图谱
本研究中,所有体系产生的次生铁矿物X射线衍射图谱出峰位置及相对强度近似一致.结合体系元素组成,参考矿物标准衍射图谱,发现本研究不同体系所得到的次生铁矿物衍射图谱中主要尖锐强峰(“J”标注)出峰位置及相对强度与标准图谱中黄铁矾类物质(黄钾铁矾:No.22-0827;黄铵铁矾:No.26-1014;草黄铁矾:No.31-0650)相关参数相一致.研究得到次生铁矿物特有的微弱宽峰(“S标注”)与标准图谱中标准施氏矿物类物质(No.47-1775)相关参数近似一致.故可以判定,本研究所有体系产生的次生铁矿物均应为黄铁矾与施氏矿物共存的混合物.
3.5 不同培养转速下Mg2+对生物合成次生铁矿物存在形态的影响
本研究中“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两处理体系Fe2+完全氧化时刻,体系次生铁矿物实际形态见图 5.由图 5可以很直观的看出,当培养转速为180 r ・ min-1且体系Fe2+完全氧化时,“Mg2+-4.8 mg ・ L-1”体系产生的约1.21 g的次生铁矿物均匀分散在培养液中.而“Mg2+-48 mg ・ L-1”体系矿物总产量为0.98 g,且约70%次生铁矿物却牢固粘附于摇瓶底部.当培养转速为100 r ・ min-1且体系Fe2+完全氧化时,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两处理体系产生的次生铁矿物均全部粘附于摇瓶底部,矿物产生量分别为0.66 g与0.74 g.
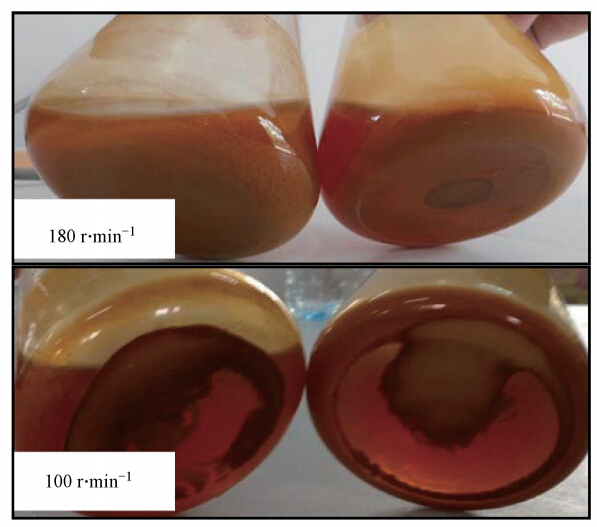
图 5 合成次生铁矿物合成体系Fe2+完全氧化时矿物的存在形态(左: Mg2+-4.8 mg ・ L-1;右:Mg2+-48 mg ・ L-1,其它元素组成与9K培养基相同)
不同体系次生铁矿物的存在状态在一定意义上决定着体系pH、Fe2+氧化率及总Fe沉淀率的变化趋势.前人研究证实,体系中存在的微生物细胞壁结构或胞外多聚物,与前期合成的次生铁矿物可以作为“晶种”加速矿物的后期合成,另外,研究表明,次生铁矿物对A. ferrooxidans存在一定量的吸附.本实验180 r ・ min-1培养条件,“Mg2+-4.8 mg ・ L-1”体系合成的矿物均匀分散于体系中,体系均匀分散的微生物菌体与前期合成的次生铁矿物均有利于后期矿物的进一步合成,进而加速体系总Fe沉淀率的增加及pH下降.而当体系Mg2+为48 mg ・ L-1时,体系合成的部分矿物不断粘附于摇瓶底部,前期合成的次生矿物及其所吸附的微生物菌体无法较好的为后期矿物合成提供模板,进而减缓体系总Fe沉淀率增加及pH的下降趋势.另外,粘附于摇瓶底部的矿物由于吸附固定一定量的A. ferrooxidans,进而减弱微生物对体系Fe2+的氧化能力,且这一推论在本研究3.2节部分结果中被很好验证.
本实验100 r ・ min-1培养条件下,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两处理体系产生的次生铁矿物均全部粘附于摇瓶底部.所以此时对体系pH、Fe2+氧化率及总Fe沉淀率变化起主导作用的不再是矿物的存在状态,而是体系Mg2+的相对含量.Mg2+是微生物细胞内酶的重要成分,亦是A. ferrooxidans生长(或其生长培养基)必需元素之一,其含量的增加可能会在一定程度上提高A. ferrooxidans活性,进而可在一定程度上提高Fe2+氧化能力及Fe3+的水解速度,进而加速总Fe沉淀.笔者认为,本研究造成次生铁矿物在摇瓶底部粘附的主要原因是初期合成的矿物与微生物菌体或微生物胞外多聚物相互作用形成包裹体,在水力剪切条件不足的情况下逐渐沉积粘附于瓶底.本研究结果已表明,培养转速越低,矿物粘附在摇瓶底部的可能性亦越大,产生次生铁矿物量相对较少.另外,由于Mg2+可在微生物胞外多聚物间形成架桥而使得微生物菌体团聚,那么Mg2+在A. ferrooxidan胞外多聚物间的架桥作用使得A. ferrooxidans菌体团聚体增大,进而使得矿物与微生物相互作用形成更大的包裹体,以致增加矿物在瓶底沉积粘附的可能性,本研究培养转速为180 r ・ min-1 条件下,“Mg2+-4.8 mg ・ L-1”与“Mg2+-48 mg ・ L-1”两处理体系矿物存在形态可较好的佐证这一推断.具体参见污水宝商城资料或http://www.dowater.com更多相关技术文档。
4 结论
1)培养转速或镁离子含量可以通过影响体系次生铁矿物存在形态及A. ferrooxidans活性来影响次生铁矿物合成体系Fe2+氧化率、总Fe沉淀率及次生铁矿物的合成量.
2)当次生铁矿物合成体系培养转速较高时,体系镁离子含量越高,体系次生铁矿物与微生物的包裹体越易粘附于反应器壁,使得体系一定量微生物由于无法游离于体系而延缓对Fe2+的氧化,同时初期合成次生铁矿物亦无法有效为后续矿物合成起到“晶种”作用,进而使得总Fe沉淀率降低,次生铁矿物合成量减少.
3)当次生铁矿物合成体系培养转速较低时,不同镁离子含量体系产生的次生铁矿物均易在反应器壁吸附,此时体系镁离子含量对微生物活性影响可能是体系Fe2+氧化率、总Fe沉淀率及矿物产生量变化的主导因素.
本研究所获相关参数及理念对生物合成次生铁矿物工艺的优化及其在酸性矿山废水治理领域应用具有一定的借鉴意义.

