1 引言(Introduction)
苯酚是一种重要的化工原料,苯酚废水主要来源于石油化工行业、陶瓷生产过程,以及以苯酚作为原料的木材厂和树脂厂等.同时,苯酚也是致癌、致畸、致变的“三致”物质,对动植物健康会造成一定的危害.因此,对苯酚废水的处理极其重要.在众多的苯酚处理方法中,非均相催化臭氧化是一种极具潜力的处理方法.
到目前为止,非均相催化臭氧氧化得到了较多的研究,但针对催化臭氧氧化的机理还未达成完全统一的观点(Nawrocki et al 2010).对催化臭氧氧化机理的研究主要存在以下3种观点:①臭氧吸附在催化剂表面,促进羟基自由基(・OH)的产生,即羟基自由基反应机理;②臭氧与有机物吸附在催化剂表面进行反应,即非均相催化剂界面效应机理;③臭氧与非均相催化剂协同作用机理.为了深入研究催化臭氧氧化苯酚的作用机理,本文主要从催化臭氧氧化机理的3种主要观点出发进行分析,采用MgO催化臭氧氧化苯酚模拟废水,借助红外分析臭氧与有机物是否在催化剂表面存在吸附;通过实验中羟基与臭氧浓度比值(Rct值)的测定计算・OH生成量,探讨MgO催化臭氧氧化降解苯酚的机理,以期为采用MgO催化臭氧氧化降解含酚废水提供一定的理论基础.
2 材料与方法(Materials and methods)2.1 实验材料
臭氧由CFJ-5型臭氧发生器现场制备,以纯度为99.99%的氧气为气源.臭氧最大浓度为85 mg・L-1,调节臭氧流量为0.1 L・min-1.采用2%的KI溶液吸收臭氧尾气.
氧化镁、氢氧化钠、苯酚、叔丁醇、对氯苯甲酸、硫代硫酸钠、碘化钾等试剂均为分析纯,由成都科龙化工试剂厂生产.
2.2 分析方法
利用高效液相色谱法测定对氯苯甲酸(pCBA),流动相分别为乙腈和pH为3的磷酸水溶液,二者之比为3∶7,流速为0.8 mL・min-1,被测样品进样量为20 μL,于波长为240 nm处进行检测.苯酚浓度的测定采用4-氨基安替比林法,臭氧浓度的测定采用靛蓝二磺酸钠比色法.
2.3 实验装置与实验方法
催化臭氧氧化苯酚实验:实验在自制的有机玻璃反应器内进行,开启臭氧发生器,调节气体流量为0.1 L・min-1,在连接反应器前预热1 min,让臭氧浓度达到恒定,实验时将预先配置好的苯酚溶液转移至反应器内,连接臭氧发生器,开始计时,每隔2 min取1次样进行分析.实验装置如图 1所示,反应器内径为50 mm,高为900 mm,有效容积为1.3 L,置于磁力搅拌器上.
叔丁醇抑制实验:选取适宜的各影响参数的量(苯酚初始浓度为100 mg・L-1,pH为7左右,温度为室温,MgO投加量为40 mg・L-1,O3投加量为3.6 mg・min-1),控制叔丁醇投加量分别为12、20、40 mg・L-1,分别在0、2、4、6、8、10、15、20 min时取样于50 mL烧杯中,加入适量硫代硫酸钠消除液相中剩余的O3,进行苯酚浓度测定.
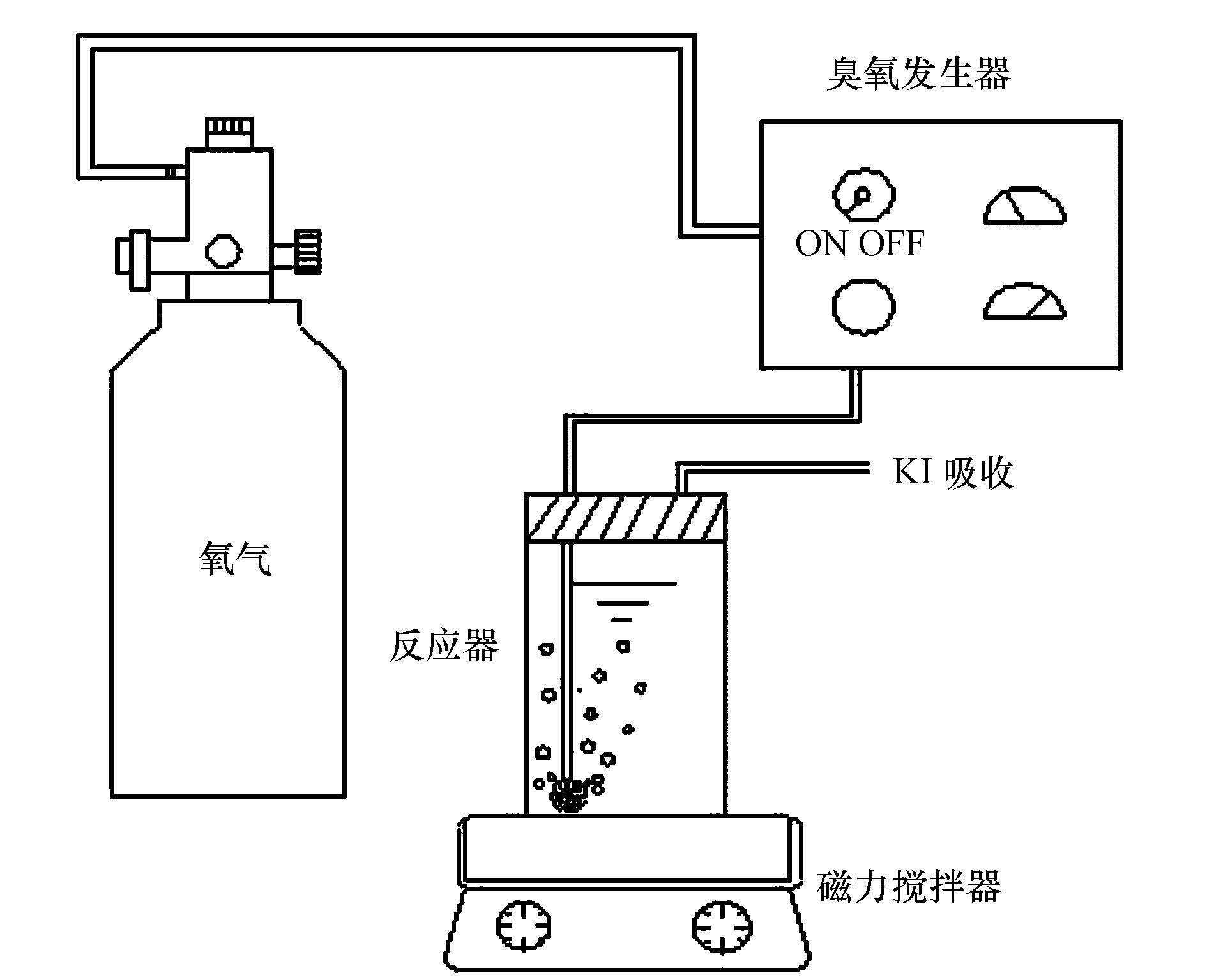
图 1实验装置图
・OH生成量测定实验:以pCBA作为・OH的捕获剂,通过测定pCBA浓度推算・OH浓度.首先,在装有900 mL纯水的烧杯中连续通入高浓度臭氧,待得到一定臭氧浓度的臭氧水后,加入100 mL溶有一定浓度苯酚和pCBA的纯水配制溶液,放于磁力搅拌器上轻微搅拌.开始计时,隔一小段时间间隔取样,分别测臭氧浓度、苯酚浓度和pCBA浓度.分别使用3根移液管在反应烧杯中直接取样,各自放置于不同的样品瓶中,其中,测定苯酚和pCBA浓度的样品瓶中事先添加适量Na2S2O3以消耗未反应的臭氧.催化臭氧氧化体系中,要加入一定质量的MgO,且取样时要先过滤.
MgO表面特性研究实验:称取80 mg MgO分别放于装有1 L纯水和1 L苯酚溶液(100 mg・L-1)的锥形瓶中,利用磁力搅拌器轻微搅拌,调节臭氧投加量为3.6 mg・min-1,通过微孔曝气头将臭氧连续通入两种体系的锥形瓶底部.开始计时,分别在反应0、2、4、6 min时,经纳米级微孔滤膜过滤适量的MgO催化剂,于30 ℃烘箱内烘数天,待其完全干燥后用于红外光谱测定.
3 结果与讨论(Results and discussion)3.1 羟基自由基抑制剂的影响
根据前人的研究,催化臭氧氧化的机理可能为:臭氧分子吸附在催化剂的表面,与表面基团发生作用,分解产生羟基自由基.为了探讨本体系是否遵循该机理,考察了在羟基自由基抑制剂存在的情况下,苯酚在催化臭氧化条件下的降解效果.
叔丁醇(TBA)是应用最为广泛的・OH抑制剂,它与・OH的反应速率常数比与臭氧的反应速率常数大很多。因此,对于遵循・OH反应机理的氧化系统,叔丁醇的存在会抑制对有机物的降解.为了证明在催化臭氧氧化体系中有・OH的产生,实验在反应过程中加入了一定量的叔丁醇,图 2为单独臭氧氧化、MgO催化臭氧氧化及・OH抑制实验的结果.由图 2可知,MgO催化臭氧氧化苯酚的降解率明显高于单独臭氧,加入叔丁醇(TBA)后MgO催化臭氧氧化苯酚明显被抑制.
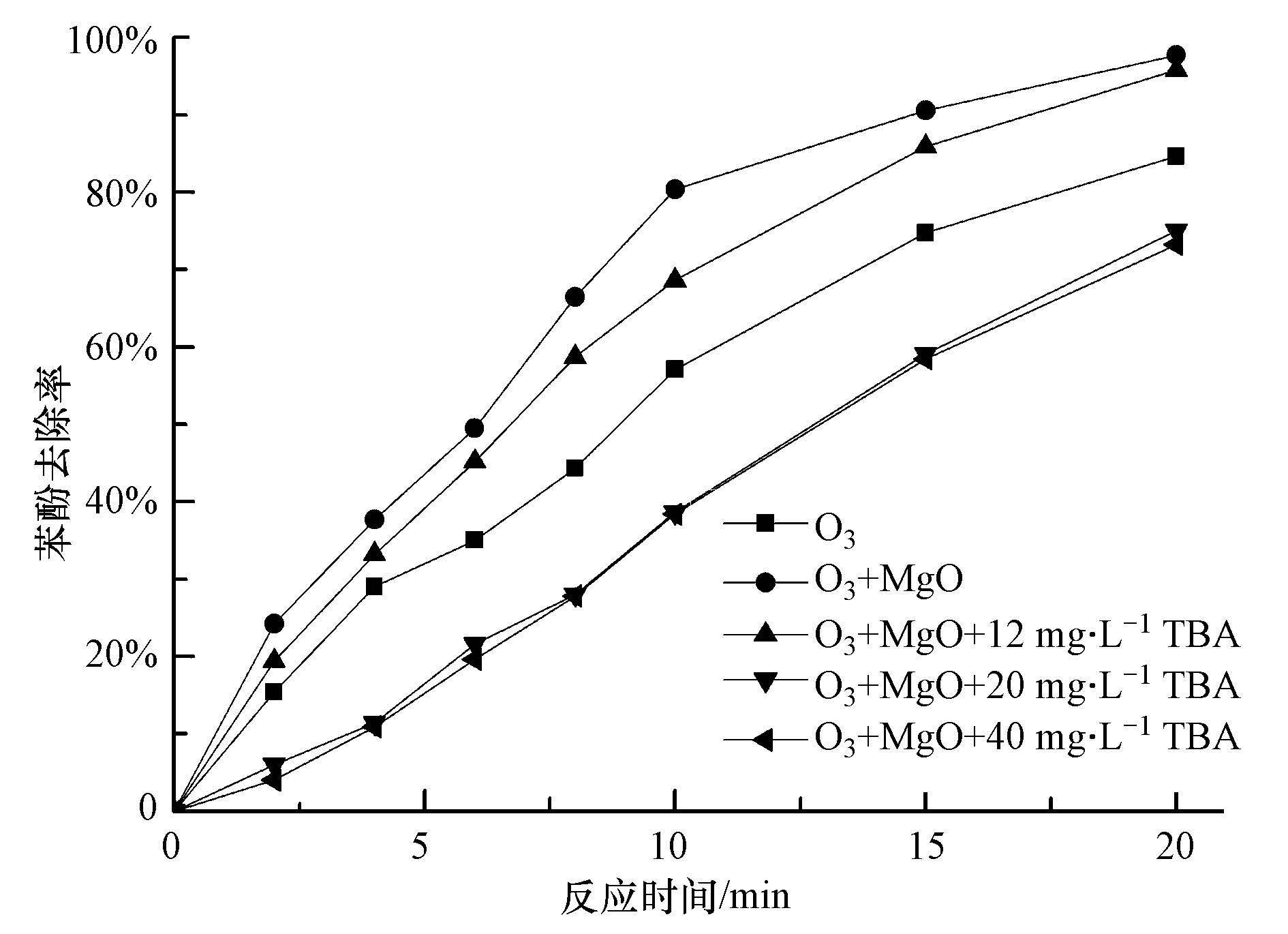
图 2不同条件下苯酚的去除效果
随着叔丁醇加入量的增加,苯酚去除率呈现先下降后保持不变的趋势.当叔丁醇加入量为12 mg・L-1时,苯酚去除率介于催化臭氧氧化和单独臭氧氧化之间;当叔丁醇加入量增加至20和40 mg・L-1时,苯酚去除率低于单独臭氧体系中苯酚的去除率.结果表明,叔丁醇的加入抑制了臭氧的链式分解,且消耗了体系中的・OH.在单独臭氧和催化臭氧氧化体系中对苯酚的去除率存在两部分:臭氧分子的氧化及・OH的氧化.当叔丁醇加入量增加至20 mg・L-1时,反应10 min时,苯酚去除率为38.6%,这部分是臭氧分子对苯酚去除率的贡献;与单独臭氧氧化相比,苯酚去除率降低了20%,这部分是单独臭氧氧化体系中・OH对苯酚去除率的贡献;同样与催化臭氧氧化相比,苯酚去除率降低了近45%,这部分是催化臭氧氧化体系中・OH对苯酚去除率的贡献.实验结果显示,在催化臭氧氧化体系中,・OH反应机理为主要的降解机理.
3.2 羟基自由基的生成量
通过上述分析可知,在单独臭氧和催化臭氧两种体系中,均有・OH的存在.为了更加明确催化臭氧氧化机理是否遵循・OH反应机理,本文对两种体系下・OH的生成量进行了测定.由于・OH在水相中的存在时间极短,很难用直接方法对其进行测定,因此,对比两种体系下的Rct值(Elovitz et al,1999)即可间接得知・OH浓度的大小关系.
根据对氯苯甲酸(pCBA)与・OH的反应速率极快(kpCBA/・OH=5.2×109L・mol-1・s-1),而与臭氧分子的反应速率慢(kpCBA/O3=0.15 L・mol-1・s-1)的特性,实验选用pCBA作为・OH的指示剂.・OH的生成量可通过pCBA 的浓度变化推导得出,如下式所示:
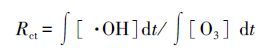
式中, ∫ [・OH]dt为・OH的总含量(mol・L-1); ∫ [O3]dt为臭氧的总含量(mol・L-1);kpCBA/・OH为pCBA与・OH的反应速率常数, kpCBA/・OH=5.2×109 L・mol-1・s-1.通过测定pCBA和O3浓度随时间的变化数据,以 ∫ [O3]dt为横坐标,ln([pCBA]/[pCBA]0)为纵坐标作图,所得曲线斜率除以pCBA与・OH的反应速率常数即可得Rct值,间接得出・OH的量.
3.2.1 臭氧浓度标准曲线的绘制
采用靛蓝二磺酸钠法来测定液相中臭氧的浓度,分别配制0、0.5、1、1.5、2、2.5 mg・L-1臭氧量的标准样于1 cm的比色皿中,在610 nm波长处利用可见光分光光度计来测定其对应的吸光度,绘制臭氧浓度标准曲线,结果如图 3所示.
由图 3可知,臭氧浓度与吸光度呈良好的线性关系,R2高达0.99757,得到的拟合曲线方程为y=0.38617x-0.02238,其中,x为臭氧浓度(mg・L-1);y为零浓度臭氧吸光度A0与各浓度臭氧吸光度A之差.
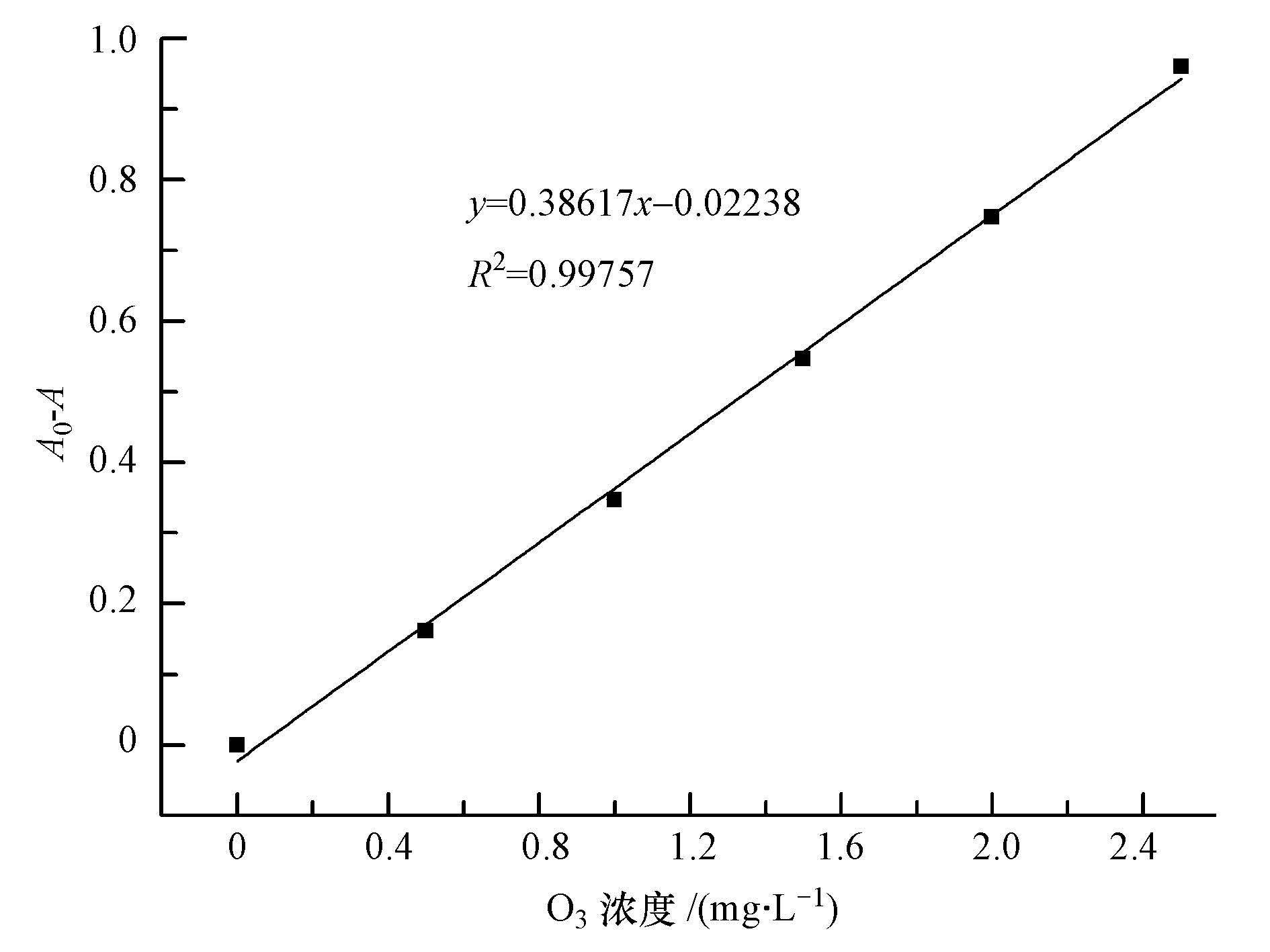
图 3臭氧浓度标准曲线图
3.2.2 pCBA标准曲线的绘制
pCBA极微溶于水,分别称取6.9、11.2、15.7、22.9 mg pCBA于1000 mL纯水中,放于磁力搅拌器上剧烈搅拌4 h,待完全溶解后利用高效液相色谱进行测定.以峰面积为横坐标,pCBA浓度为纵坐标绘图,结果如图 4所示.
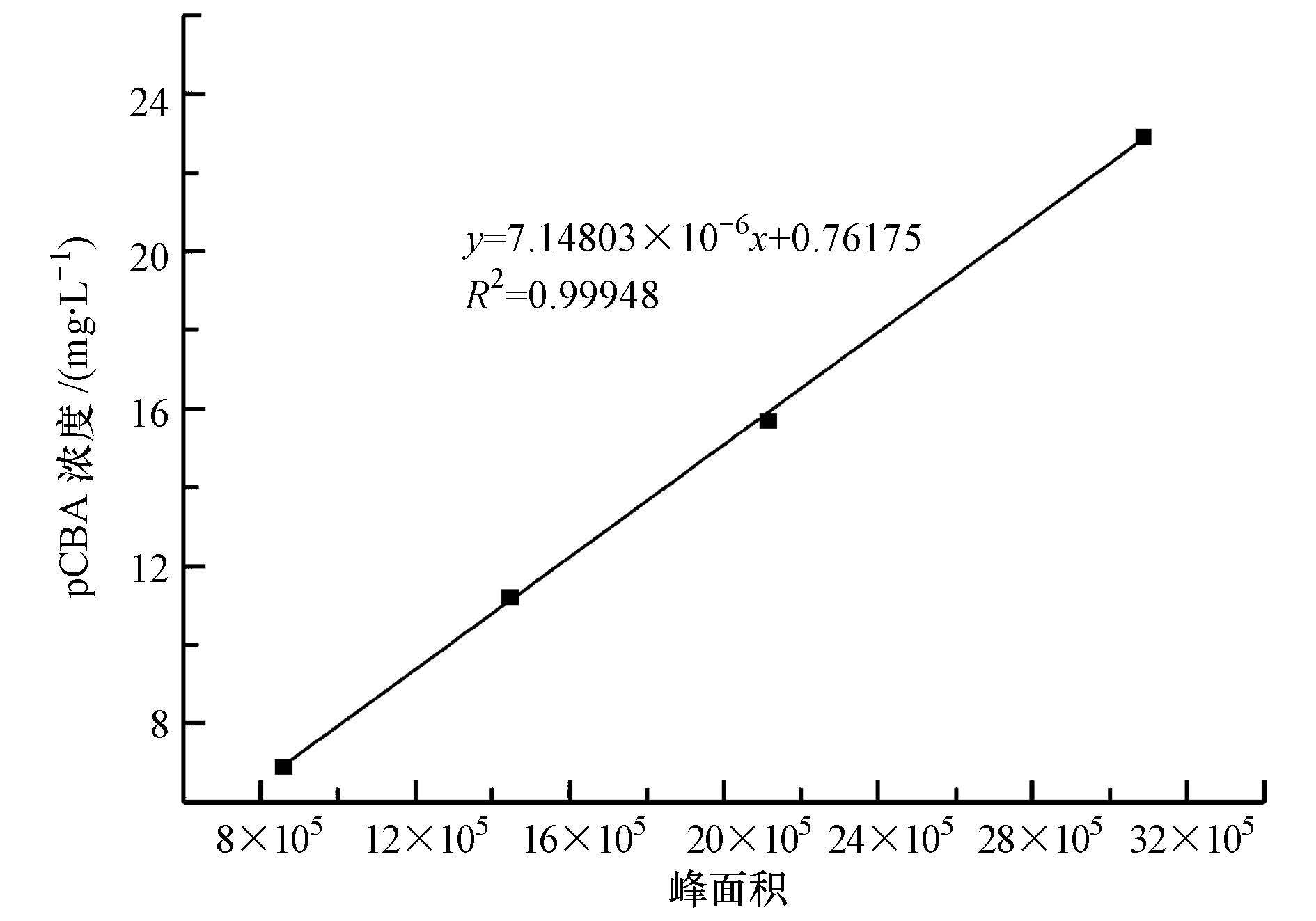
图 4 pCBA标准曲线图
由标准曲线拟合图可以看出,pCBA浓度与峰面积能够高度拟合,R2高达0.99948,pCBA标准曲线方程为:y=7.14803×10-6x+0.76175;其中,x为高效液相色谱峰面积,y为pCBA浓度(mg・L-1).
3.2.3 单独臭氧氧化体系Rct值的测定
在预先制备好的900 mL臭氧水中,加入100 mL pCBA溶液,于磁力搅拌器上轻微搅拌,开始计时,每隔1 min取样,分别测定臭氧浓度和pCBA浓度,且在测pCBA浓度的样品中加入适量的硫代硫酸钠,以消耗溶液中残余的臭氧.实验结果如图 5所示.
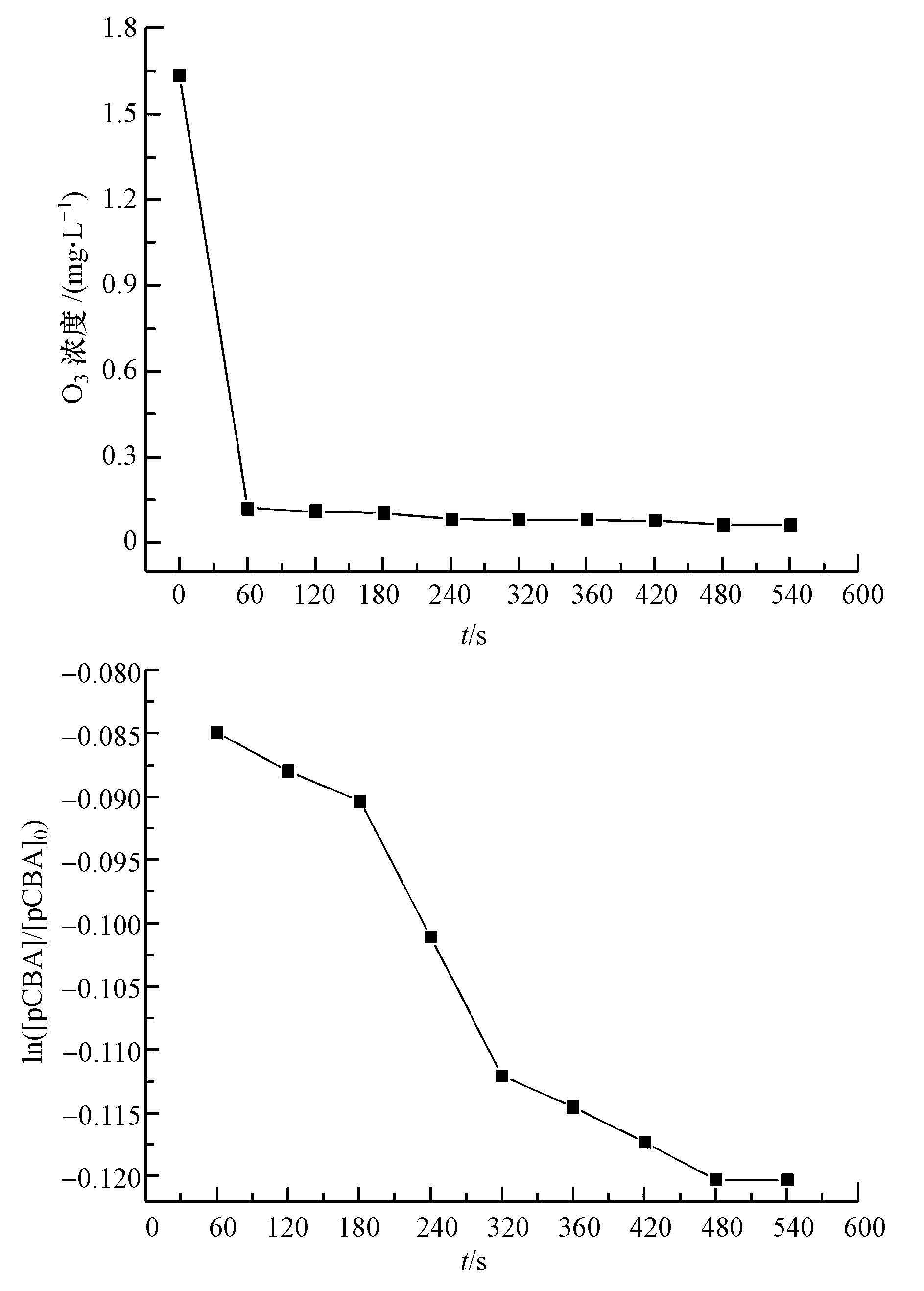
图 5单独臭氧氧化过程中O3浓度和pCBA浓度的变化
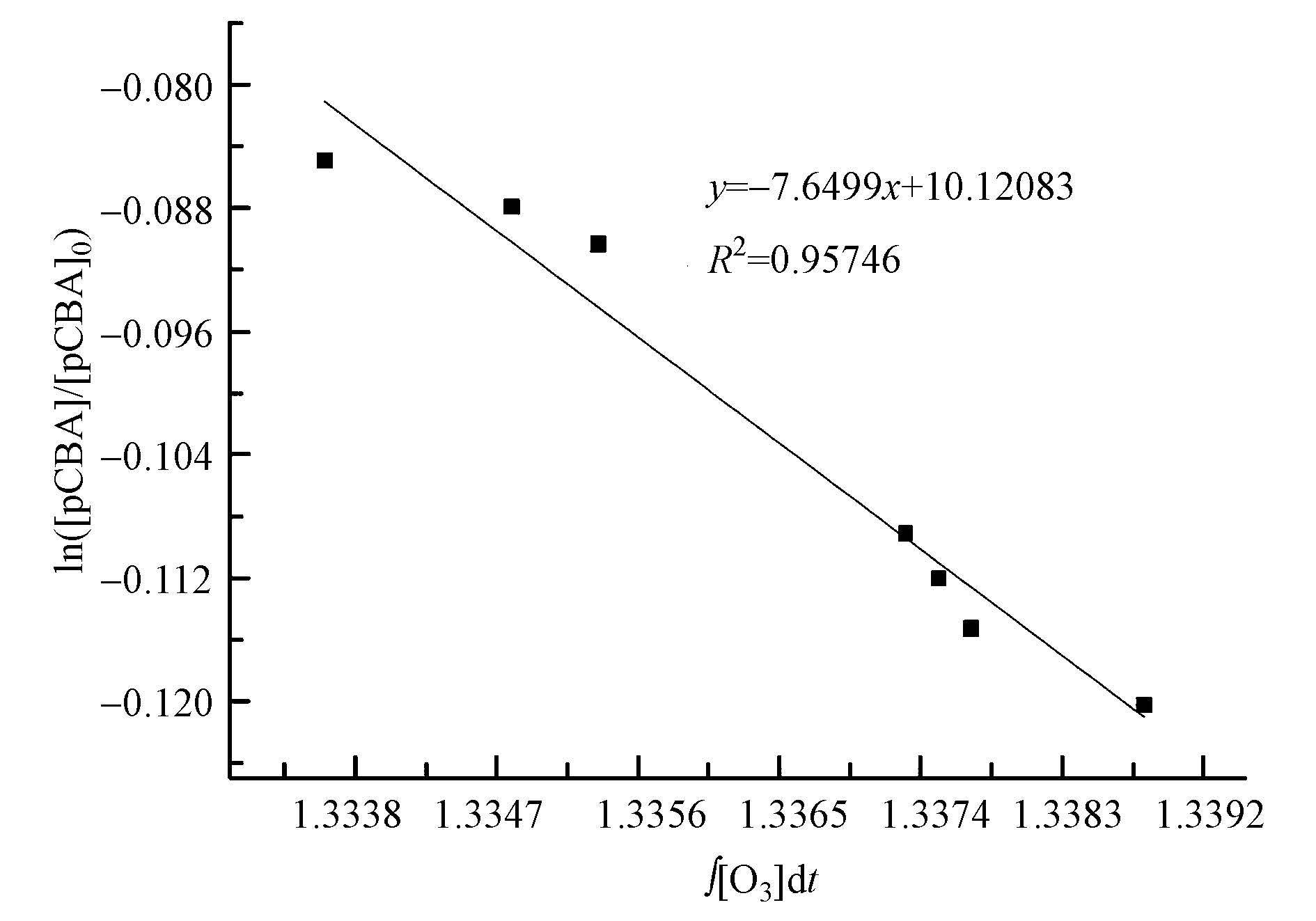
图 6单独臭氧化过程中ln([pCBA]/[pCBA]0)随 ∫ [O3]dt的变化
图 5与图 6分别为单独O3氧化过程中O3浓度和pCBA浓度随反应时间的变化,以及单独O3氧化过程中ln([pCBA]/[pCBA]0)随∫[O3]dt的变化图.由图 5可知,臭氧水中的O3浓度在1 min内快速下降,1 min后基本保持在一个较低的浓度;pCBA浓度也随时间的延长呈现出一个整体下降的趋势,表明在单独O3氧化体系中存在・OH,并且对pCBA进行了氧化降解.由图 6可知,ln([pCBA]/[pCBA]0)与 ∫ [O3]dt所得曲线的R2高达0.95以上,数据点基本在同一条直线上,曲线拟合符合规律.拟合所得直线为y=-7.6499x+10.12083,斜率为-7.6499 L・mg-1・min-1,经单位换算斜率为-6.37×103 L・mol -1・s-1,kpCBA/・OH=5.2×109 L・mol -1・s-1,经过计算可得,单独O3氧化体系中Rct值为1.47×10-9.且拟合所得直线斜率不随 ∫ [O3]dt变化,因此,斜率除以-kpCBA/・OH得到的Rct值也不随反应时间而进行变化,Rct值是单独O3氧化体系中・OH与O3任意时刻的比值.
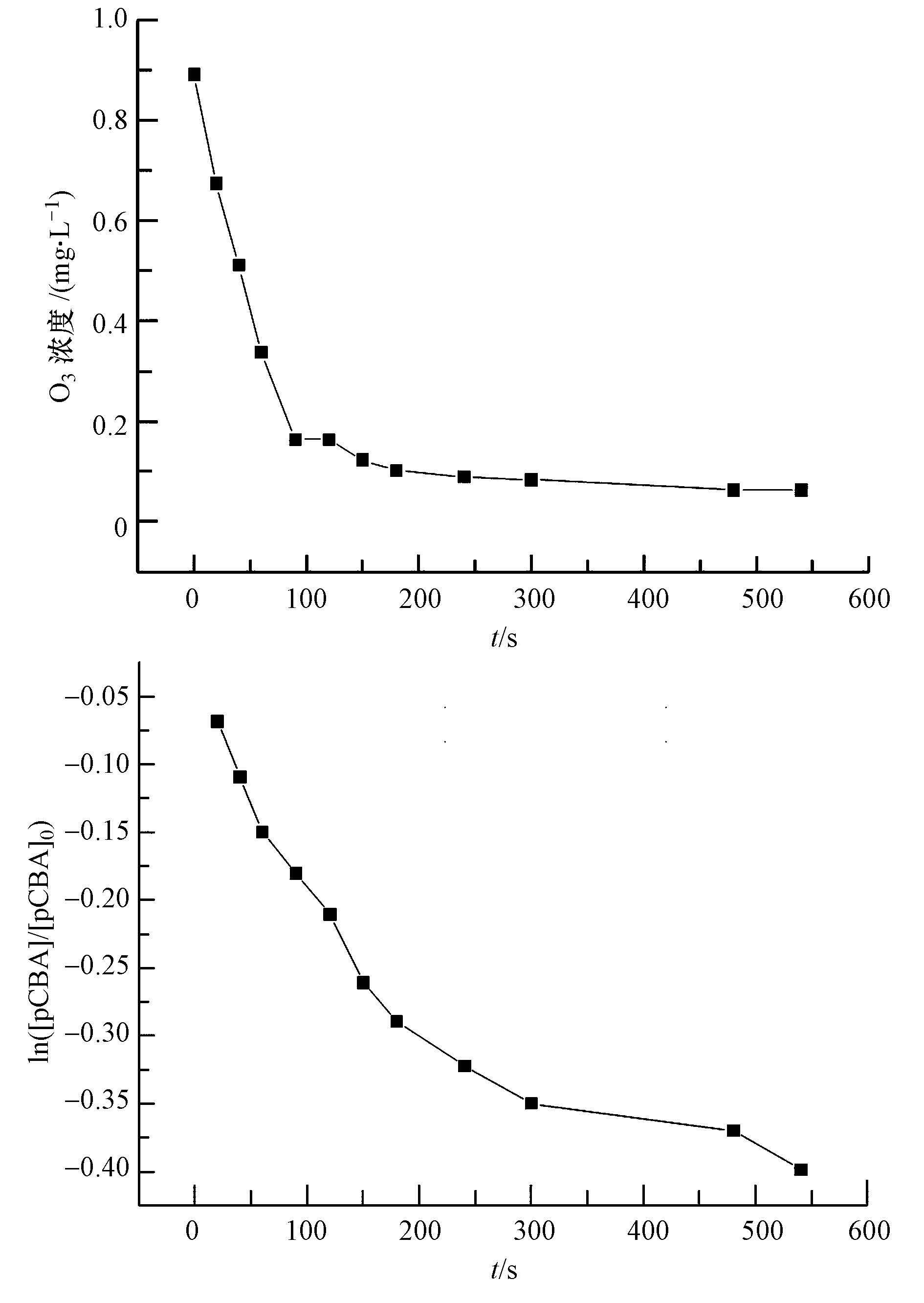
图 7 MgO催化臭氧氧化过程中O3浓度和pCBA浓度的变化
3.2.4 MgO催化臭氧氧化体系Rct值的测定
在预先制备好的900 mL臭氧水中,加入100 mL pCBA溶液及20 mg MgO催化剂,于磁力搅拌器上轻微搅拌,开始计时,分别在0 s、20 s、40 s、60 s、90 s、120 s、150 s、3 min、4 min、5 min、7 min和9 min时取样,分别测定臭氧浓度和pCBA浓度,且在测pCBA浓度的样品中加入适量的硫代硫酸钠,以消耗溶液中残余的臭氧.实验结果如图 7所示.
图 7与图 8分别为MgO催化臭氧氧化过程中O3浓度和pCBA浓度随反应时间的变化,以及MgO催化臭氧氧化过程中ln([pCBA]/[pCBA]0)随 ∫ [O3]dt的变化图.由图 7可知,臭氧浓度随时间的延长而逐渐下降,在100 s后下降速度变缓,基本保持在一个较低的浓度范围内;pCBA浓度也随时间的延长而逐渐降低,与单独臭氧氧化体系相比,降低速度有所提高.表明在催化臭氧氧化体系中pCBA被氧化速率快,体系中・OH浓度高.由图 8可知,所得拟合曲线的R2高达0.956以上,数据点基本在同一条直线上,曲线拟合符合规律.拟合所得直线方程为y=-16.3586x+6.11264,斜率为-16.3586 L・mg-1・min-1,经单位换算斜率为-13.65×103 L・mol-1・s-1,kpCBA/・OH=5.2×109 L・mol-1・s-1,经过计算可得,催化臭氧氧化体系中Rct值为3.15×10-9,是单独臭氧氧化体系中Rct值的2.14倍.且拟合所得直线斜率不随 ∫ [O3]dt变化,因此,斜率除以-kpCBA/・OH得到的Rct值也不随反应时间而进行变化,Rct值是MgO催化臭氧氧化体系中・OH与O3任意时刻的比值.
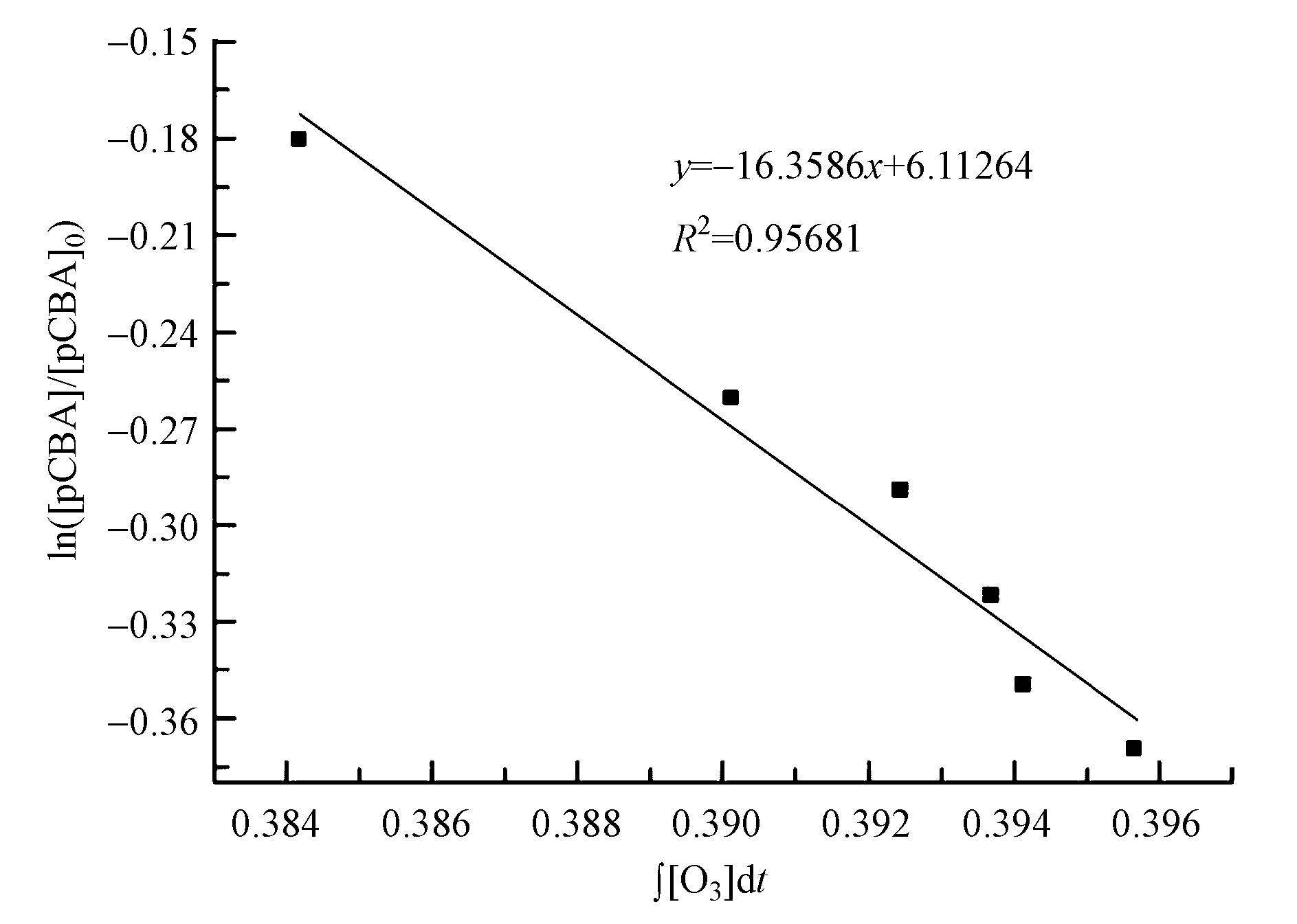
图 8 MgO催化臭氧氧化过程中ln([pCBA]/[pCBA]0)随 ∫ [O3]dt的变化
经过上述两种反应体系的Rct值测定可知,催化臭氧氧化体系中的Rct值是单独臭氧氧化体系中Rct值的2.14倍,说明在相同条件下,催化臭氧氧化体系中・OH的量是单独臭氧氧化体系中的2.14倍,进一步说明在非均相催化臭氧氧化体系中,MgO催化剂促进了臭氧分解,产生了更多的・OH,从正面验证了羟基自由基机理.
3.3 臭氧在催化臭氧体系中的存在位置
由上述实验可知,在臭氧氧化体系中,・OH确实存在,且在催化剂存在的条件下,・OH的产生量增大,催化剂促进了臭氧分解产生了更多的・OH.但催化剂促进臭氧分解产生・OH的机制还不是很明确,实验利用MgO催化剂表面性质来对这一过程进行深入研究.实验分别利用红外对MgO-纯水、MgO-苯酚废水中的MgO表面基团进行探讨.对纯水体系和苯酚体系中反应0、2、4、6 min时的催化剂进行收集过滤干燥,用以红外光谱测定,进而分析催化剂表面基团的变化,实验结果如图 9所示.
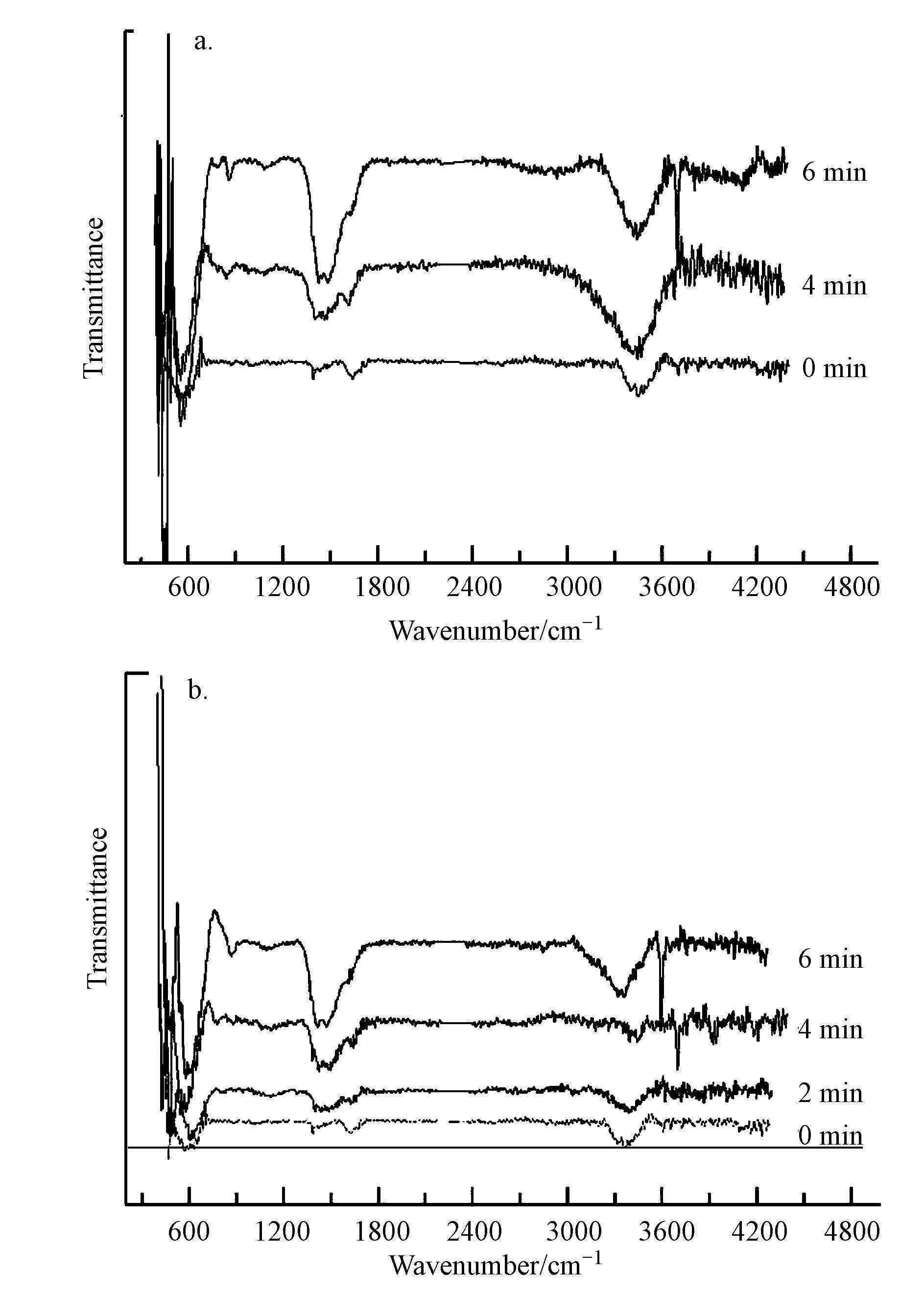
图 9 MgO-苯酚(a)和MgO-纯水(b)催化臭氧氧化体系中MgO表面基团随时间的变化
由图 9可以看出,红外光谱图中主要有3个宽吸收峰:3600~3000、1800~1200及800~400 cm-1.其中,3600~3000 cm-1和1800~1200 cm-1处存在的宽吸收峰为催化剂表面羟基吸收峰(Fuente et al,2015),800~400 cm-1处存在的峰是催化剂吸附臭氧后生成的活性氧吸附物种的峰(Fei et al,2012),具体过程如方程式(6)~(7)所示,其中,S代表催化剂.
样品在0 min时存在表面羟基吸收峰,说明MgO催化剂确实存在表面羟基,而表面羟基又是O3吸附在催化剂表面的吸附位点.在纯水体系中,随着反应时间的延长,800~400 cm-1处的峰值逐渐增大,表明臭氧在催化剂表面吸附的浓度逐渐增大.相应于3600~3000 cm-1和1800~1200 cm-1处的峰值也随时间的延长而增大,表明催化剂的表面羟基密度逐渐增大.苯酚体系中这3处的峰值也随时间而逐渐增大,且强度要高于纯水体系.
表面羟基的作用是成为催化剂的活性中心,从而改变反应物质在催化剂表面的吸附形态,并且影响反应物之间的催化反应.由此可知,臭氧首先在催化剂固有的表面羟基处吸附,在催化剂表面进行臭氧的链式分解,进而产生氧化性更强的羟基,一部分释放到溶液体系中氧化降解污染有机物,一部分留在催化剂表面增加表面羟基密度,即增加活性位点,从而使更多的臭氧吸附在催化剂表面进行分解,以此不断循环.
从以上两种体系的催化剂红外谱图分析比较中可以看出,在溶液中加入模拟有机污染物苯酚后,非均相催化臭氧氧化一段时间的催化剂在3600~3000、1800~1200及800~400 cm-1处的峰值增加幅度均要大于纯水体系.即在同等条件下,苯酚体系中的催化剂表面的臭氧吸附物种和表面羟基密度均要强于纯水体系.结合图 10可知,随着pH的增大,MgO溶液的Zeta 电位由正值变为负值,其中,Zeta 电位为0时的pH值即为它的等电点,即pHpzc为10.4左右,表明MgO为碱性催化剂.且有研究表明(Ostolska et al,2014),当溶液pH值小于金属氧化物pHpzc时,金属氧化物表面呈正电性.因此可以认为,在MgO催化臭氧氧化苯酚过程中,MgO为碱性催化剂,而苯酚为弱酸性物质;且MgO表面呈正电性,苯酚在水中会发生可逆性水解,产生带负电的离子,两者之间会存在静电吸引使彼此靠近,在这个过程中,会使溶液中形成很强的能量对流,溶解于溶液中的臭氧则被更多地与催化剂发生碰撞,进而更多地被吸附在催化剂表面进行分解.因此,苯酚在非均相催化臭氧氧化过程中充当了促进臭氧吸附到催化剂表面的促进剂.总体来看,两种体系中均是臭氧吸附到催化剂表面进而分解产生羟基自由基,而且苯酚体系中的羟基自由基的产生量应较纯水体系中的多,该分析结论与上述实验结果相一致.
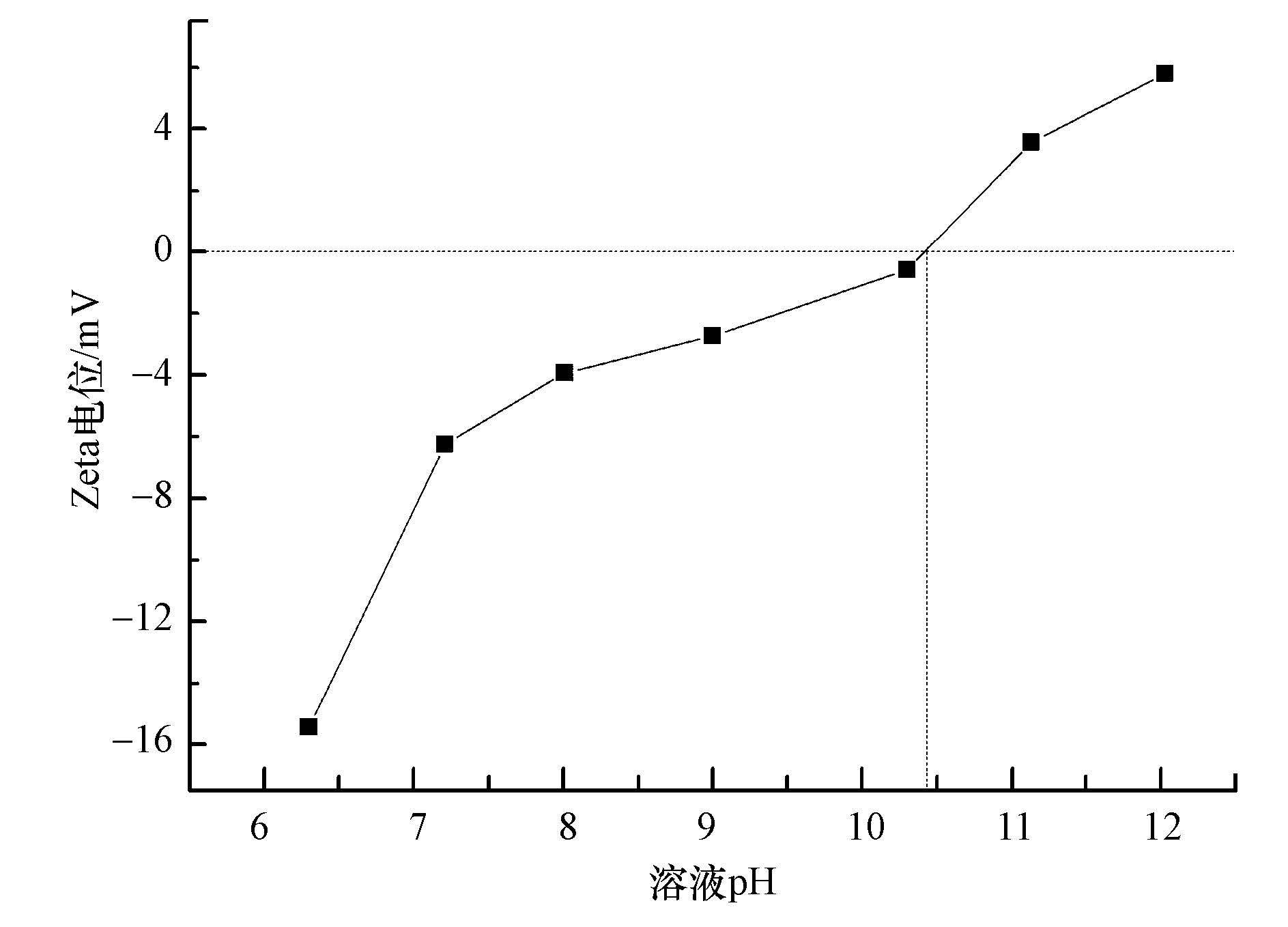
图 10 MgO溶液在不同pH值下的Zeta电位
4 结论(Conclusions)
1) MgO/O3与单独O3两种反应体系中都存在・OH.在催化臭氧氧化体系中加入40 mg・L-1的叔丁醇,反应10 min后,与不加叔丁醇时催化臭氧氧化和单独臭氧氧化相比,苯酚去除率分别降低了45%和20%.具体参见污水宝商城资料或http://www.dowater.com更多相关技术文档。
2) MgO/O3体系中的・OH是单独O3体系中的2.14倍.单独臭氧氧化和催化臭氧氧化体系中的Rct值分别为1.47×10-9和3.15×10-9.
3) 苯酚在MgO催化臭氧氧化体系中起到了促进了臭氧吸附在MgO表面的作用.红外分析得出,对苯酚进行催化臭氧氧化后,MgO催化剂表面的臭氧吸附物种峰和表面羟基峰均要高于纯水中的MgO.臭氧吸附到MgO表面后,分解产生・OH,一部分释放在溶液中降解苯酚,一部分则留在其表面增加表面羟基密度.

